West Coast Retina
Case of the Month
March, 2011
A 31-year-old male with fuzzy vision in his left eye.
Presented by Sandeep Randhawa, MD

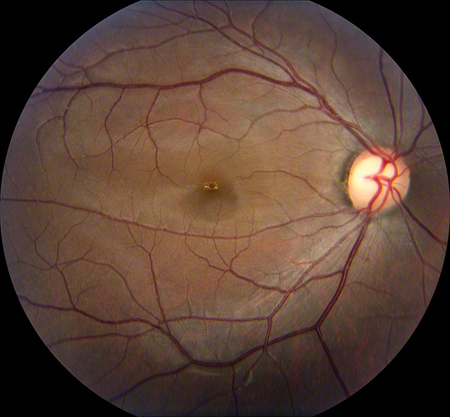
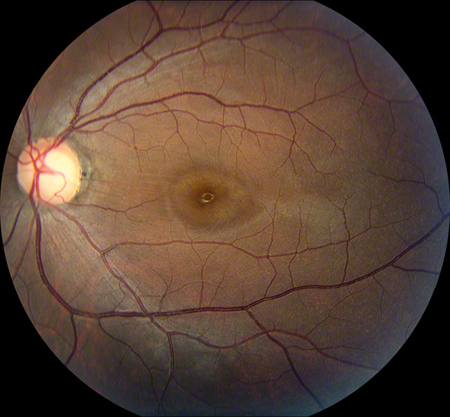
Figures 1: Color fundus photographs showing a mild foveal pigmentary change in the right macula and a bull’s eye lesion (central hyperpigmentation surrounded by a ring of hypopigmentation) in the left macula.
Case History
A 31-year-old Chinese male presented to the clinic reporting decreased vision in his left eye which he first noticed a week prior to presentation. He denied any flashes, floaters, recent trauma, or history of eye surgery. He had borderline hypertension and his past ocular history only included moderate myopic astigmatism. His father had been diagnosed with a macular hole in the past. He denied any nyctalopia or photopsia.
On examination, best-corrected visual acuity was 20/32 in the right eye and 20/50 in the left. Intraocular pressure, confrontation visual field, and anterior segment examination were normal in both eyes.
Dilated fundus exam showed a mild foveal pigmentary change in the right macula, but the left macula revealed changes showing central hyperpigmentation surrounded by a ring of hypopigmentation (a bull’s-eye lesion). There was attenuation of the foveal reflex and significant macular pigmentary changes in the left eye. There was slight temporal pallor of the optic discs bilaterally. The vessels and periphery were normal both eyes (Figure 1).
What is your Diagnosis?
Differential Diagnosis
A 31-year-old Chinese male presents with acutely symptomatic decrease in vision in his left eye with a ipsilateral bulls eye maculopathy.
The differential diagnosis of a bull’s-eye maculopathy includes choroquine/hydroxychloroquine toxicity, Stargardt’s disease, central areolar choroidal atrophy, chronic macular hole, fenestrated sheen macular dystrophy, benign concentric annular macular dystrophy, cone dystrophy, and cone-rod dystrophy. Other causes also include olivopontocerebellar atrophy and ceroid lipofuscinosis, but they are associated with systemic findings.
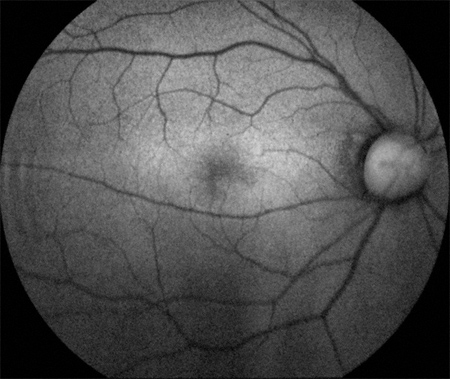
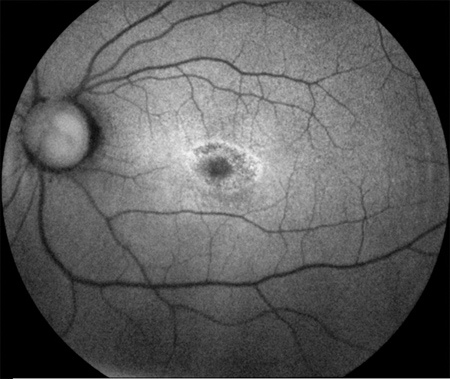
Figures 2: Fundus autofluorescence images showing a slight increase in background autofluorescence in the right parafoveal area, and a parafoveal ring of hyperautofluorescence surrounding an area of relative hypoautofluorescence at the fovea in the left eye (in a bull’s eye pattern).
Diagnosis
A Humphrey visual field test (24-2) revealed non-specific changes in the right eye and a central scotoma in the left eye. Fundus autofluorescence images showed a slight increase in background autofluorescence in the right parafoveal area, and a parafoveal ring of hyperautofluorescence surrounding an area of relative hypoautofluorescence at the fovea in the left eye (consistent with a bull’s-eye pattern, Figure 2). Fluorescein angiogram revealed a normal macula and periphery in the right eye, but the left eye revealed mild parafoveal hyperfluorescence with stippled hyperfluorescent spots in the periphery (Figure 3).
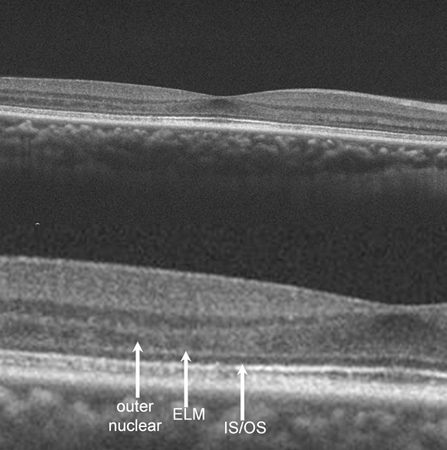
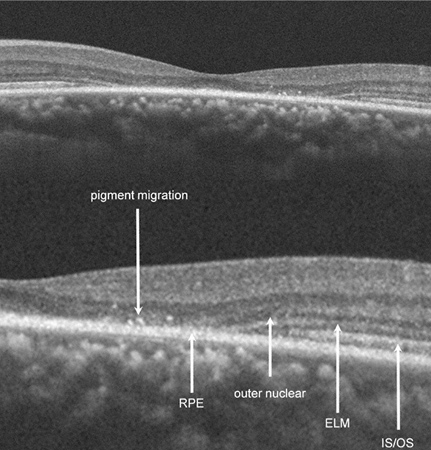
Spectral Domain OCT revealed disruption of the photoreceptor inner-segment/outer-segment junction, external limiting membrane, and outer nuclear layer in the left eye and appear to collapse into an intact RPE with an adjacent area of pigment migration. Interestingly, OCT imaging of the asymptomatic right eye also had some revealing findings: the foveal contour was noted to be abnormally shallow temporally as a result of a decrease in the outer nuclear layer thickness in this area. In addition, the photoreceptor inner-segment/outer-segment as well as the outer-segment/RPE junction demonstrated an irregularly hypo-reflective and spiculated appearance beneath the areas of outer nuclear layer thinning (Figure 4). Based on appreciation of these findings, it became clear that our patient had an asymmetric, yet bilateral disease process (asymmetric) that was more advanced in his left eye.
Based on the above, our patient was thought to have bilateral, albeit asymmetrical, photoreceptor dystrophy (worse in the symptomatic left eye) with a predominant involvement of the cones. An electroretinogram was obtained. The photopic cone response and the 30 Hz flicker response (for cones) demonstrated abnormal decreased amplitudes and increased implicit times in both eyes, more pronounced in the symptomatic left eye, but certainly present in the right eye as well (Figure 5). In addition, the ERG scotopic rod response demonstrated decreased rod response in both eyes indicating widespread rod degeneration bilaterally.
Based on the exam and the investigations, the patient was diagnosed with cone-rod dystrophy in both eyes.
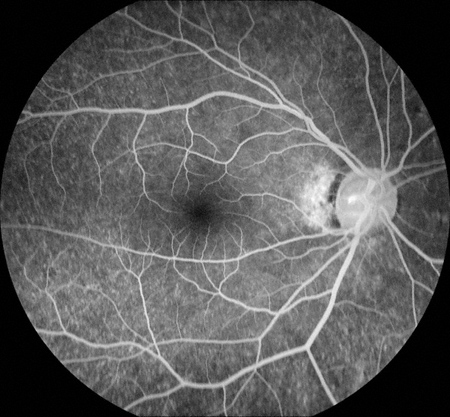
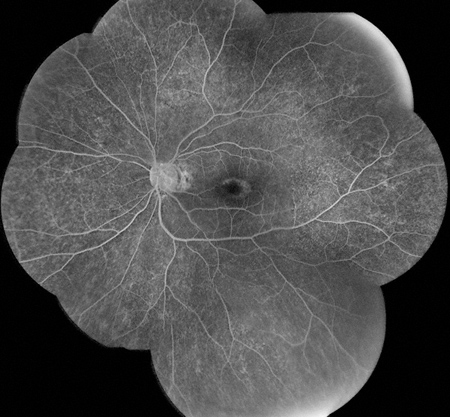
Figures 3: Fluorescein angiogram showing a normal right eye. Note the mild parafoveal hyperfluorescence with stippled hyperfluorescent spots in the periphery in the left eye.
Right Eye
Left Eye
Figures 4: Spectral domain OCT of the right eye showing an abnormally shallow foveal contour temporally as result of a decrease in the outer nuclear layer in the area. Also, the photoreceptor inner-segment/outer-segment as well as the outer-segment/RPE junction demonstrated an irregularly hypo-reflective and spiculated appearance beneath the areas of outer nuclear layer thinning. Spectral domain OCT of the left eye revealed disruption of the photoreceptor inner-segment/outer-segment junction, external limiting membrane, and outer nuclear layer in the left eye as they collapse into an intact RPE with an adjacent area of apparent pigment migration.
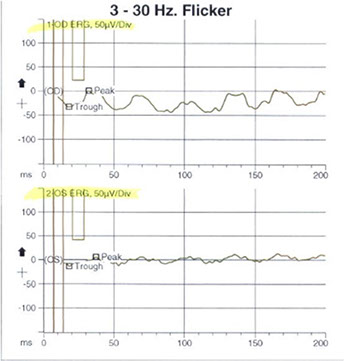
Figures 5: Electroretinogram photopic cone response and the 30 Hz flicker response (for cones) demonstrating abnormal decreased amplitudes and increased implicit times in both eyes, more pronounced in the symptomatic left eye. In addition, the scotopic rod response demonstrated decreased rod response in both eyes indicating widespread rod degeneration bilaterally.
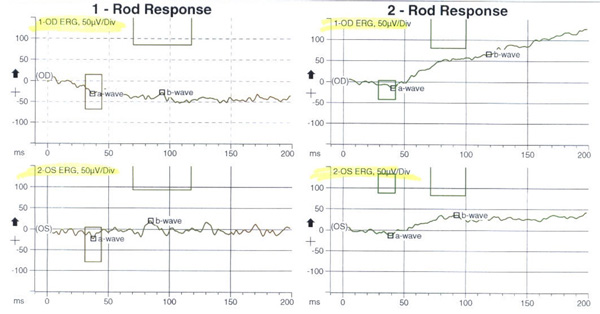
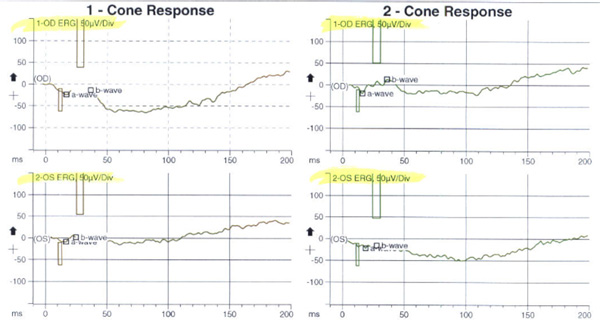
Discussion
The cone-rod dystrophies constitute a relatively rare group of cone disorders with an annual prevalence of 1/40,000.1
The earliest symptom is a decrease in the visual acuity, which is usually gradual in the first and second decade of life and may be accompanied by photophobia or hemeralopia. This is usually followed by a gradual, variable dyschromatopsia. The fundus abnormalities may be absent or subtle in the early stages which can lead to a frequent delay in diagnosis. 2-4
In contrast to the symptoms of the rod-cone dystrophies (RCDs, typical retinitis pigmentosa) resulting from predominant rod involvement, i.e., night blindness and loss of peripheral vision, the clinical signs of CRDs reflect the predominant involvement of cones, which leads to decreased visual acuity and loss of sensitivity in the central visual field, with nyctalopia occurring only in the late stages of the disease.3,4
Visual field testing shows central scotomas, while the periphery is spared. Fundus examination shows pigment deposits and various degrees of retinal atrophy in the macular region. Retinal vessels are usually normal or moderately attenuated. The optic disc is often pale at early stages, particularly on the temporal side.3,4
Fluorescein angiography and fundus autofluorescence show that the peripheral retina is also involved with heterogeneity in the fluorescence, but to a lesser extent than the macula. Secondly, the electroretinogram (ERG) shows a shift in implicit time of cone responses, followed by a decrease in both cone and rod responses. Cone responses are more severely affected than rod responses, as in our case.3
Histopathological studies of cone rod dystrophy cases have revealed reduction in the photoreceptors and fragmentation of outer segments.5
This case is particularly interesting because our patient presented with only symptoms in his left eye, and the findings on fundus examination and fluorescein angiography were markedly asymmetric. The Spectral domain OCT images contributed significantly to narrowing our differential by demonstrating a bilateral process involving the photoreceptors, and likely a photoreceptor dystrophy. This prompted the electroretinogram which confirmed the diagnosis of a cone-rod dystrophy.
Take Home Points
Cone-rod dystrophies may present asymmetrically with the patient being symptomatic only in one eye with the fundus abnormalities in the uninvolved eye being very subtle.
Autofluorescence imaging provides useful insight into the metabolic state of the photoreceptor/retinal pigment epithelium complex, serving as a useful diagnostic and prognostic tool.
Spectral domain OCT can be really helpful in delineating the pathological process and picking up subtle abnormalities which aid in the diagnosis. Although an ERG is still the gold-standard for diagnosis of cone-rod dystrophies, SD-OCT contributes to the diagnosis and allows in vivo visualization of structural photoreceptor abnormalities.
Want to Subscribe to Case of the Month?
References
- Hamel CP, Griffoin JM, Bazalgette C, Lasquellec L, Duval PA, Bareil C, Beaufrere L, Bonnet S, Eliaou C, Marlhens F, Schmitt-Bernard CF, Tuffery S, Claustres M, Arnaud B: [Molecular genetics of pigmentary retinopathies: identification of mutations in CHM, RDS, RHO, RPE65, USH2A and XLRS1 genes]. J Fr Ophtalmol 2000,23(10):985-995.
- Krill AE: Hereditary Retinal and Choroidal Diseases, Vol. 2, Clinical Characteristics. Hagerstown MD:, Harper & Row, 1977:359.
- Hamel CP. Orphanet J Rare Dis. 2007 Feb 1;2:7. Cone rod dystrophies.
- Fu AD, Ai E, McDonald HR, Johnson RN, Jumper JM. Hereditary Macular Dystrophies. In: Tasman W, Jaeger EA, eds. Duane’s Clinical Ophthalmology, Philadelphia: Lippincott Williams and Wilkins, 2005;9:1-40.
- Gregory-Evans K, Fariss RN, Possin DE, Gregory-Evans CY, Milam AH. Abnormal cone synapses in human cone-rod dystrophy. Ophthalmology 1998 Dec;105(12):2306-12.