West Coast Retina
Case of the Month
April, 2011
Presented by Jon Wender, MD
An asymptomatic 26-year-old Caucasian male with a pigmented lesion in the right macula.
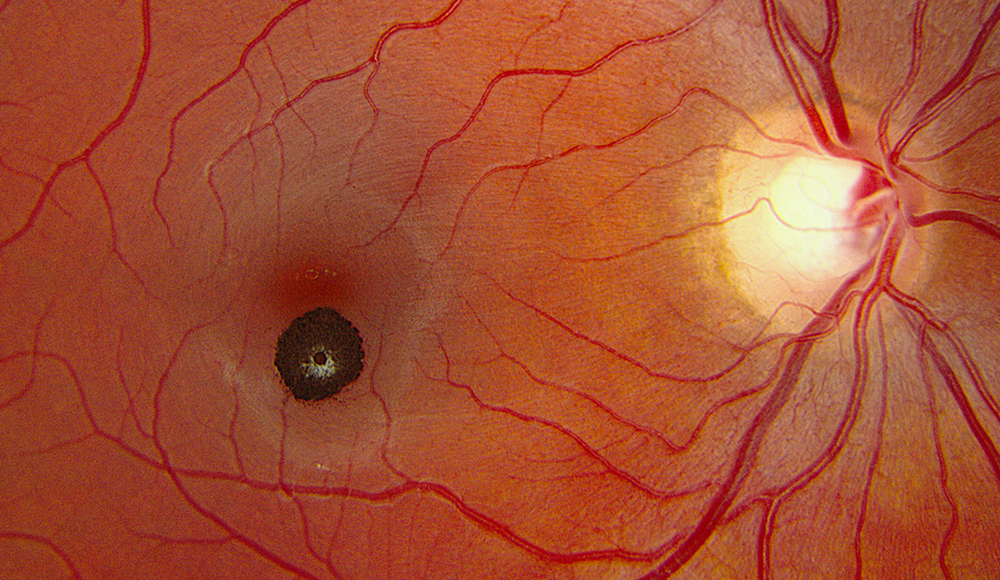
Figure 1: Color fundus photograph of the right eye demonstrating a small, pigmented, minimally elevated lesion located inferior to the fovea and associated with sharp borders and whitish overlying tissue.
Case History
A 26-year-old Caucasian man was referred for a macular lesion incidentally discovered on routine examination of the right eye. His past ocular history was non-contributory and his past medical history was significant for gastroesophageal reflux disease. He did not take any medications. The review of systems was negative.
On examination, visual acuity was 20/13 in each eye. Intraocular pressure and the anterior segment examination were normal in each eye. The dilated fundus examination of the right eye revealed a small, well-circumscribed, darkly pigmented, minimally elevated mass located just inferior to the fovea and associated with a small amount of overlying white-gray tissue (Figure 1). The lesion was not associated with any hemorrhage, exudate, macular edema, subretinal fluid, or atrophy of the retinal pigment epithelium (RPE).
Fluorescein angiography (FA) revealed early hypofluorescence secondary to blockage and late hyperfluorescence surrounding and overlying the well-demarcated lesion (Figure 2). Spectral domain optical coherence tomography (SD-OCT) revealed elevation and hyperreflectivity of the inner surface of the lesion (Figure 3). Optical shadowing and well-defined tumor margins were seen on OCT as well. The retina and RPE surrounding the lesion were normal.
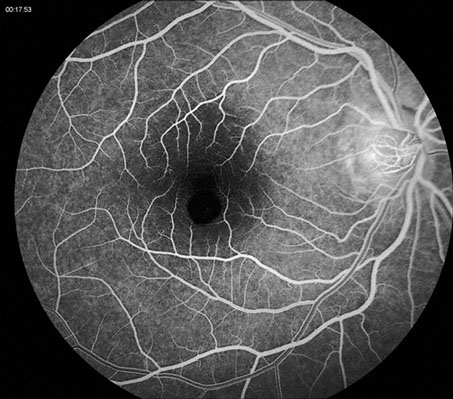
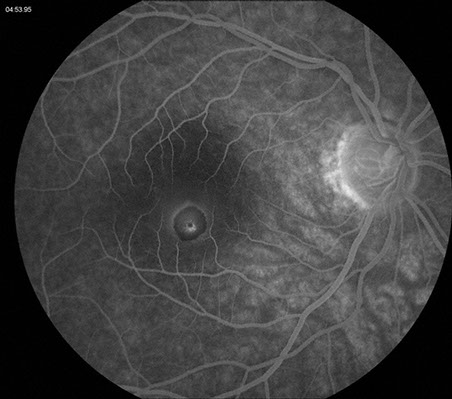
Figures 2: Fluorescein angiography of the lesion of the right macula. Note the early hypofluorescence secondary to blockage from the increased pigmentation. A slight halo of late hyperfluorescence is seen as well as some increased fluorescence centrally.
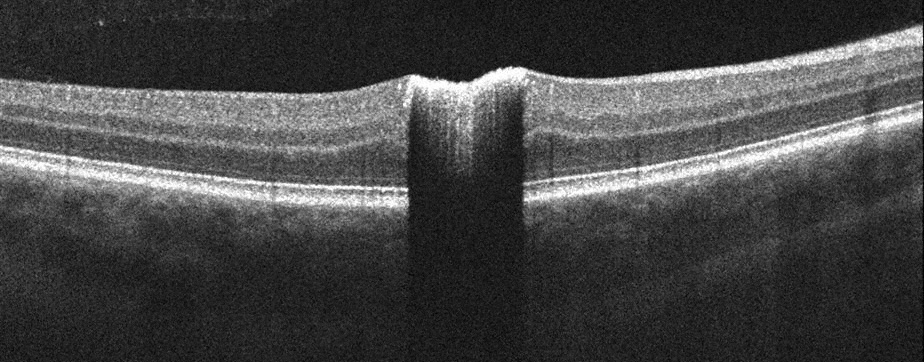
Figure 3: Spectral domain optical coherence tomography of the right macula. A horizontal scan through the lesion shows increased reflectivity, sharp tumor margins, and significant optical shadowing.
What is your Diagnosis?
Differential Diagnosis
The differential diagnosis of congenital simple hamartoma of the RPE includes congenital hypertrophy of the RPE (CHRPE), pigmented epiretinal membrane (ERM), acquired RPE hyperplasia, intraretinal foreign body, RPE adenoma, RPE adenocarcinoma, and retinal invasion by a choroidal melanoma or nevus.3,7The clinical, angiographic, and OCT features of this darkly-pigmented, well-demarcated, minimally elevated, hyperreflective macular lesion were consistent with a diagnosis of congenital simple hamartoma of the RPE.
Discussion
A hamartoma is defined as “a benign tumor-like malformation resulting from faulty development in an organ and composed of an abnormal mixture of tissue elements that develop and grow at the same rate as normal elements but are not likely to compress adjacent tissue.”1
Congenital simple hamartoma of the RPE, described by Laqua in 1981,2 is a rare, benign, darkly-pigmented, discrete, full-thickness retinal mass that typically occurs in close proximity to the fovea.3 The lesion has been reported to occur in Caucasians, Hispanics, Asians, and Africans.3-5 It is thought to be congenital in nature as it may be diagnosed in childhood and is not progressive. The tumor has not been found to be associated with systemic abnormalities.3
Patients with congenital simple hamartoma of the RPE are usually asymptomatic and visual acuity is typically normal unless the lesion is subfoveal6 or there is significant macular traction.3 The tumor tends to be small in size, with a basal diameter of less then 1.5 mm and an average thickness of 1.6mm.3,5 Minimally dilated feeder and drainage vessels3, as well as retinal hemorrhage7, may sometimes be present but intraretinal and subretinal fluid are typically lacking.3, 4 The retina and RPE surrounding the tumor are normal.8
Fluorescein angiography typically reveals blockage by the tumor and a mild overlying or surrounding rim of hyperfluorescence in the later phase. Optical coherence tomography of the lesion often demonstrates abrupt margins, slight retinal elevation, and hyperreflectivity associated with significant optical shadowing.6,8 Vitreomacular adhesion in the area of the tumor may also be seen on OCT in some cases.6 Indocyanine green angiography may reveal a dark area secondary to blockage of the underlying choroidal fluorescence by the tumor.7 B-scan ultrasonography may demonstrate a nodular tumor configuration and increased reflectivity.3
It is important to distinguish congenital simple hamartoma of the RPE from a pigmented ERM because an attempt to surgically remove a full-thickness retinal tumor could yield a very unfavorable outcome. A distinction from ERM can be made based on a younger age at presentation and complete optical shadowing on OCT, which are seen in congenital simple hamartoma of the RPE.6
Malignant transformation has not been reported in any cases of congenital simple hamartoma of the RPE and multiple publications have shown this lesion to be non-progressive over extended periods of follow up.3,5,7 The recommended management, therefore, should be conservative, involving photographic documentation and serial observation.
Take Home Points
- Congenital simple hamartoma of the RPE is a rare, benign tumor that is typically located close to the fovea and is not associated with any systemic conditions.
- The lesion tends to be small and darkly pigmented. Although retinal hemorrhage may rarely occur, intraretinal and subretinal fluid are typically absent.
- Fluorescein angiography may reveal blockage of choroidal fluorescence by the tumor with a ring of late hyperfluorescence. Optical coherence tomography of the tumor typically demonstrates hyperreflectivity and optical shadowing.
- The differential diagnosis includes CHRPE, pigmented ERM, RPE hyperplasia, choroidal melanoma, choroidal nevus, RPE adenoma, RPE adenocarcinoma, and intraretinal foreign body.
- The lesion is typically non-progressive and management should be conservative.
Want to Subscribe to Case of the Month?
References
- The American Heritage Medical Dictionary: Houghton Mifflin, 2007.
- Laqua H. Tumors and tumor-like lesions of the retinal pigment epithelium. Ophthalmologica 1981;183(1):34-8.
- Shields CL, Shields JA, Marr BP, et al. Congenital simple hamartoma of the retinal pigment epithelium: a study of five cases. Ophthalmology 2003;110(5):1005-11.
- Madgula IM, Adatia FA, Sagoo MS, Wescott M. Simple hamartoma of the retinal pigment epithelium in a man of African descent. Can J Ophthalmol 2009;44(4):e35-6.
- Gotoh M, Yoshikawa H, Kagimoto HT, Ishibashi T. Congenital simple hamartoma of the retinal pigment epithelium in an Asian. Jpn J Ophthalmol 2008;52(2):144-5.
- Shukla D, Ambatkar S, Jethani J, Kim R. Optical coherence tomography in presumed congenital simple hamartoma of retinal pigment epithelium. Am J Ophthalmol 2005;139(5):945-7.
- Lopez JM, Guerrero P. Congenital simple hamartoma of the retinal pigment epithelium: optical coherence tomography and angiography features. Retina 2006;26(6):704-6.
- Shields CL, Materin MA, Karatza EC, Shields JA. Optical coherence tomography of congenital simple hamartoma of the retinal pigment epithelium. Retina 2004;24(2):327-8.