West Coast Retina
Case of the Month
April, 2013
Presented by Sara Haug, MD, PhD
A 40-year-old Indian man presents with decreased vision in his right eye.

A
B
C
D
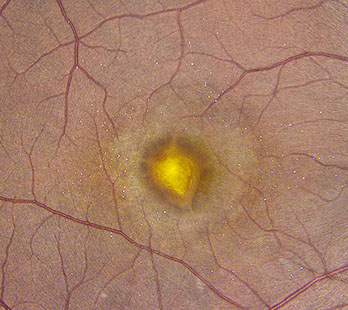
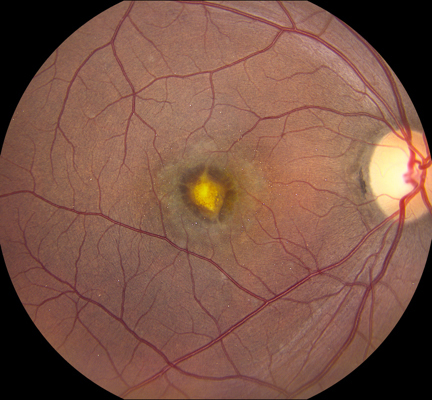
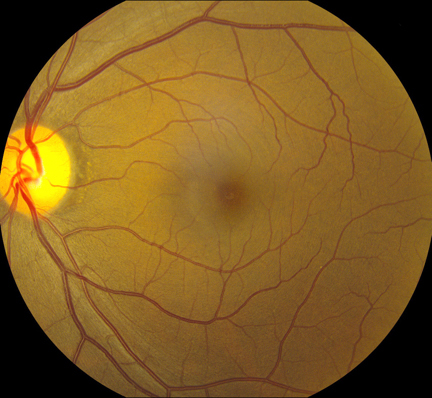
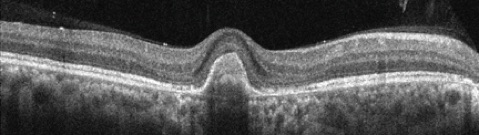
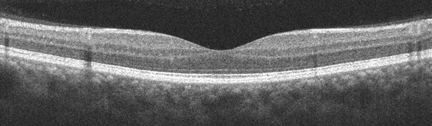
Figures 1A-E: Color fundus photograph of the right and the left macula with accompanying Optical Coherence Tomography (SD-OCT) images. The right macula (Figs. 1A and E) shows a yellowish submacular lesion surrounded by a ring of pallor. Highly refractile crystals are noted on the retinal surface. These are best seen on the magnified view below (Fig 1E). SD-OCT of the right eye shows a subretinal pigment epithelial lesion and the hyper-reflective crystals on the posterior hyaloid face and inner surface of the retina (Fig 1B). The left macula appears unremarkable with normal OCT findings (Figs 1C & D).
E
Case History
A 40-year-old Indian man, recently relocated from Ohio, presented with decreased vision in his right eye. Past history was significant for unilateral multifocal choroiditis complicated by choroidal neovascularization (CNV) in the right macula. Cataract extraction with posterior chamber intraocular lens placement in the right eye had been recently performed. Otherwise, past medical and surgical histories were unremarkable. The family history was non-contributory.
Best-corrected vision was 20/40 on the right and 20/16 on the left. Intraocular pressure was 13 mmHg on the right and 10 mmHg on the left with open angles on gonioscopy. The blood pressure was 116/67. There was no afferent pupillary defect. Anterior segment examination on the right showed a well-centered posterior chamber intraocular lens. Anterior segment examination of the left eye showed a mild nuclear sclerotic cataract. Posterior segment examination of the right eye showed a central subretinal scar with a surrounding annular area of retinal pigment epithelial atrophy and scattered highly refractile crystals on the surface of the retina (Figures 1A & E). Posterior segment examination of the left eye was unremarkable (Figure 1C).
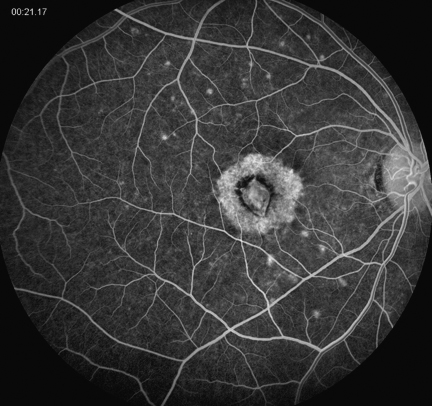
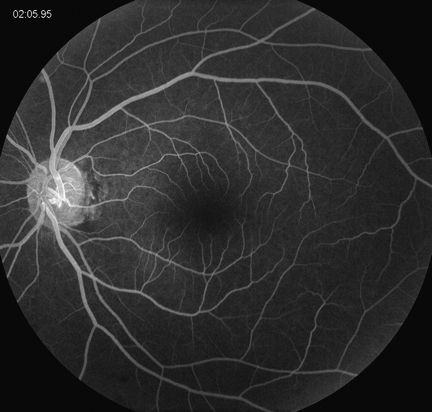
Spectral domain optical coherence tomography (SD-OCT) of the right eye showed a subretinal pigment epithelial mass and hyperreflective crystals that localized to the posterior surface of the hyaloid as well as the inner surface of the retina (Figure 1B). Fluorescein angiography of the right eye revealed a window defect corresponding to the area of RPE atrophy as well as scattered small window defects consistent with healed areas of choroiditis. There was no evidence of active choroidal neovascularization. (Figure 2A) The SD-OCT and fluorescein angiogram of the left eye were normal. (Figures 1C and 2B)
A
B
Figures 2A and B: Fluorescein angiography of the right and left macula. In the right eye, note the hyperfluorescent staining of a subfoveal scar surrounded by pigment epithelial atrophy and multiple small hyperfluorescent lesions (Fig 2A). The angiogram of the left eye is normal (Figure 2B).
What is your Diagnosis?
Differential Diagnosis
The differential diagnosis for crystalline retinopathy is large and includes toxic exposure, metabolic disorders, genetic diseases, vascular diseases and idiopathic causes.1 Toxic exposure from canthaxanthine, tamoxifen, nitrofurantoin, intravitreal triamcinolone and secondary hyperoxaluria due to methoxyflurane or ethylene glycol ingestion can cause crystalline retinopathy.1,2 Metabolic disorders resulting in crystalline retinopathy include secondary hyperoxaluria caused by excessive rhubarb ingestion or hyperabsorption of oxalate. Several conditions can lead to the hyperabsorption of oxalate including sarcoidosis, liver cirrhosis, small bowel resection, and renal failure.1 Genetic causes of retinal crystalline deposition are Bietti’s crystalline dystrophy, gyrate atrophy, cystinosis, Sjogren-Larrson syndrome and primary hereditary hyperoxaluria.1 Vascular conditions in which one might see retinal crystals include idiopathic juxtafoveal telangiectasia and talc retinopathy.1,3 Finally, there are several idiopathic conditions that can lead to crystalline retinopathy, including chronic retinal detachment (usually associated with retinal dialysis), white dot fovea syndrome and West African crystalline maculopathy (secondary to Kola nut ingestion).1,4,5
Clinical Course
Our patient was a healthy man with no known metabolic or genetic disorders. He had no history of retinal detachment or toxic exposure and denied intravenous drug use. The patient was not from West Africa. Further history obtained from the patient’s ophthalmologist in Ohio described a submacular CNV that had been previously treated with intravitreal injections of bevacizumab, pegaptanib and photodynamic therapy combined with intravitreal triamcinolone injection. A diagnosis of crystalline retinopathy secondary to intravitreal triamcinolone injection was made.
Discussion
Triamcinolone-associated crystalline retinopathy is a novel syndrome of crystalline maculopathy associated with a history of intravitreal triamcinolone injections. The crystals have been described as refractile, multi-colored, located in the posterior pole, and unassociated with obvious visual deficit or retinal sequelae.2
Triamcinolone is a corticosteroid with low solubility in aqueous solution. The proposed mechanism for retinal crystal formation is aggregation of the insoluble components of triamcinolone. In the report by Sarraf et al., resolution of the crystalline retinopathy was observed following vitrectomy surgery with membrane peeling. Given the OCT data showing the crystals to localize to the inner retinal surface and posterior hyaloid face, this is an expected result.2
Take Home Points
- Intravitreal triamcinolone can cause a crystalline retinopathy with no adverse visual effects or retinal sequelae.
- Vitrectomy surgery with membrane peeling results in resolution of the retinal crystals.
Want to Subscribe to Case of the Month?
References
- Drenser K, Sarraf D, Jain A, Small KW. Crystalline Retinopathies. Surv Ophthalmol 2006;51:535-49.
- Sarraf D, Vyas N, Jain A, et al. Triamcinolone-associated crystalline maculopathy. Arch Ophthalmol 2010;128:685-90.
- Sallo FB, Leung I, Chung M, et al. Retinal crystals in type 2 idiopathic macular telangiectasia. Ophthalmology 2011;118:2461-7.
- Rajak SN, Mohamed MD, Pelosini L. Further insight into West African crystalline maculopathy. Arch Ophthalmol 1984;102:584-7.
- Ahmed I, McDonald HR, Schatz H, et al. Crystalline retinopathy associated with chronic retinal detachment. Arch Ophthalmol 1998;116:1449-53.