West Coast Retina
Case of the Month
August, 2013
A 23 year-old man with decreased vision in both eyes
Presented by Steven Williams, MD

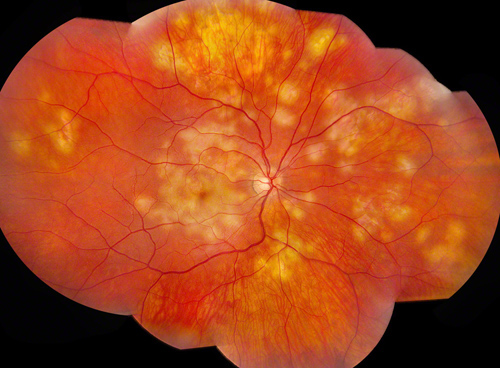
A
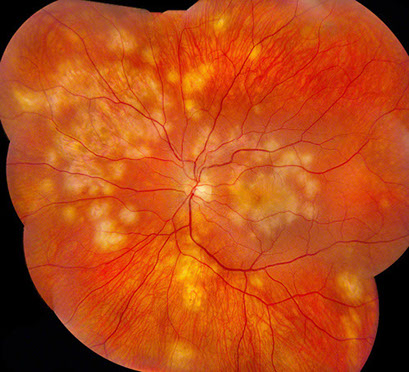
B
Figures 1A and B: Color montage fundus photographs of both eyes demonstrate creamy ovoid chorioretinal lesions, most densely concentrated in the macula and mid-peripheral retina.
Case History
A 23 year-old Caucasian, male college student presented with decreased vision for one week with the left eye more severely affected than the right eye. His past ocular history was notable for moderate myopia. Family history was significant for glaucoma (paternal grandfather) and corneal transplantation (both grandmothers). He denied any recent flu-like illness or history of sexually transmitted diseases.
Vision in the right eye was 20/20-2 and 20/250-4 on the left eye. Intraocular pressure was normal bilaterally. Anterior segment examination showed 1+ anterior chamber cell in the right eye and 2+ cell in the left eye. There were scattered cells in the anterior vitreous of both eyes. Posterior segment examination showed multiple areas of chorioretinal scaring and multiple chorioretinal lesions involving the macula and mid peripheral retina, consistent with active chorioretinitis bilaterally (Figures 1A and B).
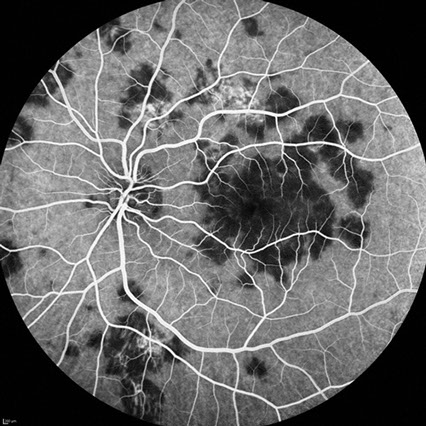
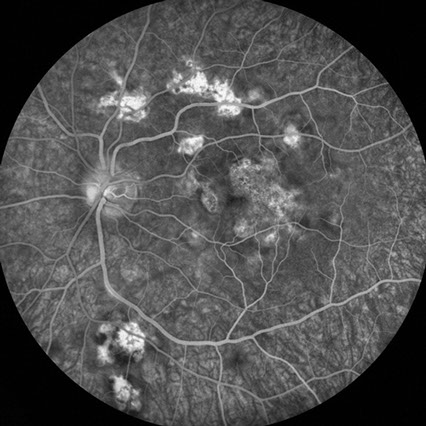
Spectral domain OCT revealed patchy disruption of the retinal pigment epithelium (RPE) and photoreceptor outer segments, associated with areas of outer retinal hyperreflectivity and thinning of the outer plexiform layer. Shallow subretinal fluid was also present (Figure 2A). The inner retinal layers were intact (Figures 2A and B). Fundus autoflourescence showed multiple geographic areas of hypoautoflourescence, surrounded by a rim of relative hyperautofluorescence (Figures 3A and B). Fluorescein angiography demonstrated early hypofluorescence of the chorioretinal lesions, followed by late hyperfluorescence, without leakage (Figures 4A and B). Indocyanine green (ICG) angiography reveled early hypofluorescence of the placoid chorioretinal lesions with blocking of underlying choroidal perfusion. These lesions showed persistent hypofluorescence peripherally, with mild central hyperfluoresence in the late phase (Figure 5A and B).
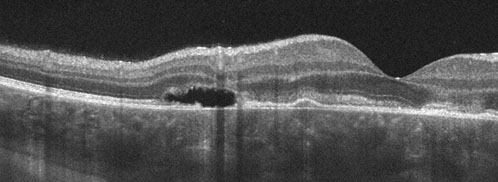
A
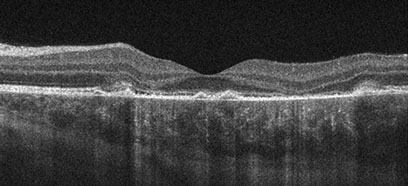
B
Figures 2A and B: Spectral domain OCT of the macula, demonstrates focal areas of RPE elevation and atrophy, associated with disruption and loss of overlying photoreceptor outer segments. There are also small pockets of subretinal fluid. Focal hyper-reflectivity of the photoreceptor IS/OS junction is also noted. There is patchy loss of the outer plexiform layer, though the inner retinal layers remain intact.
A
B
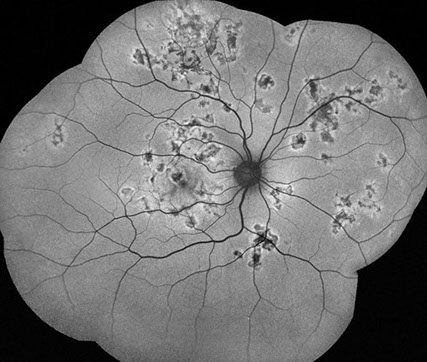
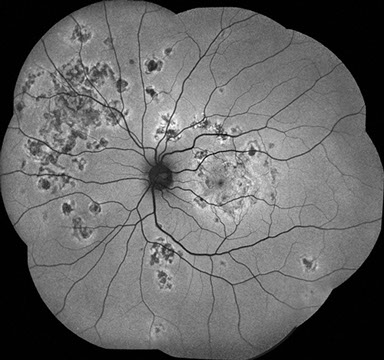
Figures 3A and B: Fundus autoflourescence shows multiple geographic areas of hypoautoflourescence, surrounded by a rim of relative hyperautofluorescence.
A
B
Figures 4A and B: Fluorescein angiography of the left eye reveals early hypofluorescence, followed by late staining of the placoid chorioretinal lesions.
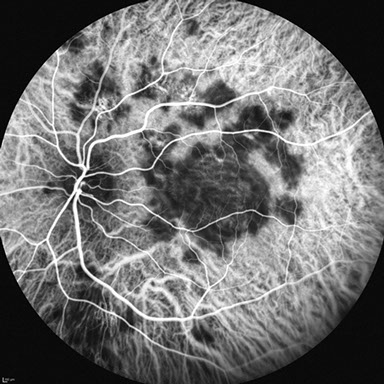
A
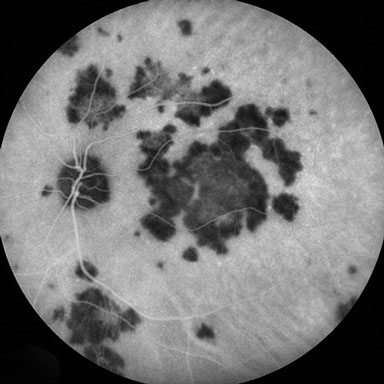
B
Figures 5A and B: Indocyanine green angiography of the left eye reveals early hypofluorescence of the placoid lesions, which shows mild central hyperfluorescence in the late phase. The chorioretinal lesions are associated with blockage of choroidal fluorescence.
What is your Diagnosis?
Differential Diagnosis
Diagnostic considerations for chorioretinitis includes infectious entities such as Mycobacterium tuberculosis, syphilis and necrotizing herpetic retinitis. Inflammatory conditions such as serpiginous chorioretinopathy, acute posterior multifocal placoid pigment epitheliopathy (APMPPE), sarcoidosis and birdshot chorioretinopathy are also included. The APMPPE-serpiginous chorioretinopathy continuum also includes related disorders such as macular serpiginous, relentless placoid chorioretinitis, and persistent placoid maculopathy. Intraocular lymphoma is also a consideration.
Discussion
Laboratory investigations for syphilis, tuberculosis, and sarcoidosis were unrevealing and the patient was diagnosed clinically with APMPPE. Acute posterior multifocal placoid pigment epitheliopathy (APMPPE), was first described by Gass in 1968. The disease typically affects young adults, often resulting in rapid vision loss in one or both eyes from macular involvement by multiple placoid chorioretinal lesions. Lesions are usually flat, well circumscribed, postequatorial, and gray-white or yellow in appearance. The lesions begin to fade within a few days of onset of symptoms and may result in extensive RPE alterations. Vitritis is seen in roughly 50% of patients, subretinal fluid is seen in 30-40% of patients, and a flu-like prodrome is reported by one-third of patients. Less common ocular findings include perivenous exudation, papilledema, papillitis, optic neuritis, iridocyclitis, and cerebral or systemic vasculitis. Both eyes are usually affected, though onset may differ by weeks or months.
Acutely in the disease, flourescein angiography shows early blocking of choroidal filling by the chorioretinal lesions, followed by mid- and late-phase staining of the lesions. As the lesions resolve, large choroidal vessels may become more visible over depigmented or atrophic RPE. ICG angiography demonstrates choroidal hypofluorescence correlating to blockage by overlying chorioretinal lesions. The areas of hypofluoresence decrease as the disease resolves. Fundus autofluoresence can show hypo- and hyperautofluoresence depending on the disease course and the degree of RPE atrophy. There is no evidence of choroidal atrophy. Spectral domain OCT typically shows disruption of the RPE and photoreceptors outer segments. Hypereflectivity from the outer plexiform layer to the RPE is also observed and usually fades over the course of weeks to months. Choroidal neovascularization and recurrence of the disease are seen infrequently. Electroretinogram and electro-oculogram are typically normal during the acute phase of the disease. While many patients describe a viral prodrome, the cause the disease remains unknown. The disease is usually self-limited, although short-term treatment with systemic corticosteroids has been used in some cases. Visual prognosis is generally good, as most eyes retain 20/30 vision or better. Small, persistent scotomas are not uncommon.
Clinical Course
Our patient was treated with a short course of systemic prednisone and vision improved to 20/20 in the right eye and 20/25 in the left eye within two weeks of initiating therapy. His chorioretinal lesions resolved leaving areas of RPE atrophy (Figures 6A and B).
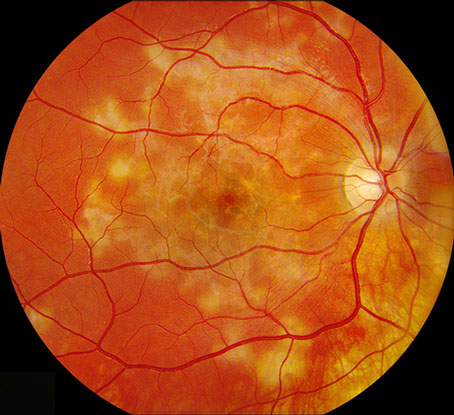
A
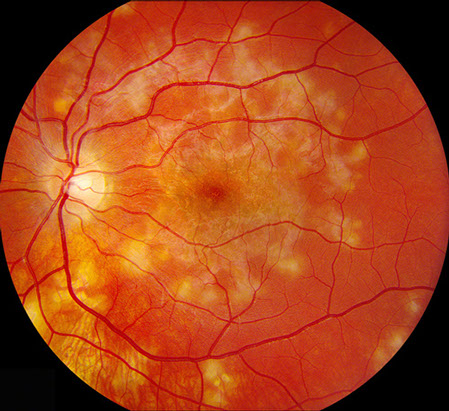
B
Figures 6A and B: Fundus photographs of the right and left eye show resolving chorioretinal lesions with RPE alterations throughout the macula.
Take Home Points
- APMPEE is an acute, bilateral chorioretinitis that tends to affect young adults and is characterized by early blocking and late staining of chorioretinal lesions on fluorescein angiography.
- The disease is usually self limited and visual prognosis is good
- Spectral domain OCT can demonstrate a variety of findings in APMPPE, including disruption of the photoreceptors and RPE, hyperreflectivity of the outer retinal layers and subretinal fluid.
Want to Subscribe to Case of the Month?
References
- Gass JD. Acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol. 1968;80(2):177-85.
- O’Halloran HS et al. Acute multifocal placoid pigment epitheliopathy and central nervous system involvement: nine new cases and a review of the literature. Ophthalmology. 2001;108(5):861-8.
- Hsu CT, Harlan JB, Goldberg MF, Dunn JP. Acute posterior multifocal placoid pigment epitheliopathy associated with a systemic necrotizing vasculitis. Retina. 2003;23(1):64-8.
- Jones BE et al. Relentless placoid chorioretinitis: A new entity or an unusual variant of serpiginous Chorioretinitis? Arch Ophthalmol. 2000;118(7):931-8.
- Kovach JL. Persistent placoid maculopathy imaged with spectral domain OCT and autofluorescence. Ophthalmic Surg Lasers Imaging. 2010;41 suppl:S101-3.
- Quillen DA et al. The white dot syndromes. Am J Ophthalmol. 2004;137(3):538-50.
- Scheufele TA, Witkin AJ, Schocket LS et al. Photoreceptor atrophy in acute posterior multifocal placoid pigment epitheliopathy demonstrated by optical coherence tomography. Retina. 2005;25(8):1109–1112.
- Fiore T el al. Acute posterior multifocal placoid pigment epitheliopathy: outcome and visual prognosis. Retina. 2009;29(7):994-1001.
- Birnbaum AD, Blair MP, Tessler HH, Goldstein DA. Subretinal fluid in acute posterior multifocal placoid pigment epitheliopathy. Retina. 2010;30(5):810–4.
- Lee GE, Lee BW, Rao NA, Fawzi AA. Spectral domain optical coherence tomography and autofluorescence in a case of acute posterior multifocal placoid pigment epitheliopathy mimicking Vogt-Koyanagi-Harada disease: case report and review of literature. Ocul Immunol Inflamm. 2011;19(1):42-7.
- Steiner S, Goldstein DA. Imaging in the diagnosis and management of APMPPE. Int Ophthalmol Clin. 2012;52(4):211-9.
- Yeh S et al. Fundus autofluorescence imaging of the white dot syndromes. Arch Ophthalmol. 2010;128(1):46-56.