West Coast Retina
Case of the Month
December, 2014
Presented by Steven Williams, MD
A 40-year-old Chinese woman with one year history of reduced adaptation to dim lighting

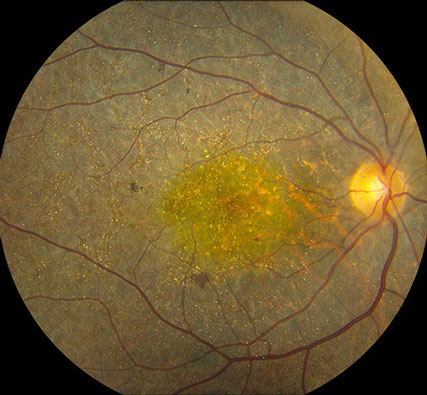
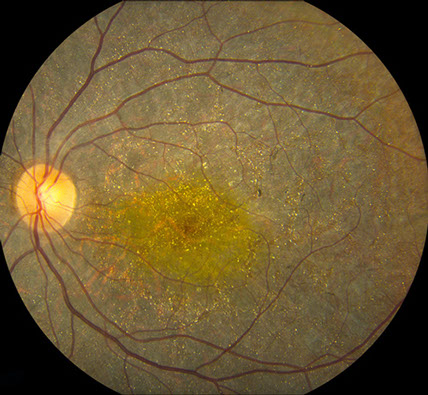
Figure 1: Fundus photograph of the right and left eye. Note the widespread crystals and areas of retinal pigment epithelial atrophy. The optic nerve and retinal vessels have a normal appearance
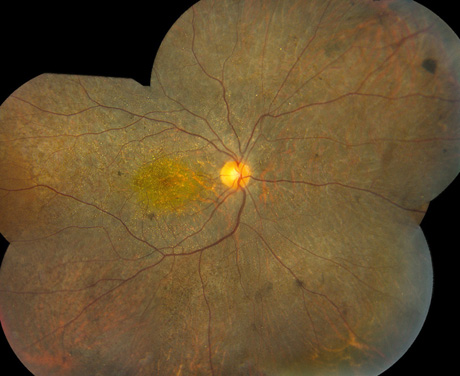
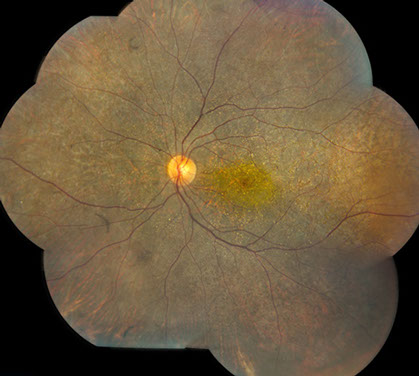
Figure 2: Fundus photograph montage of right and left eye demonstrating diffuse crystalline retinopathy as well as areas of RPE hyperplasia and pigment migration. There is also marked chorioretinal atrophy in the posterior pole revealing the choroidal vasculature.
Case History
A 40 year old, Chinese woman presented with a one-year history of poor adaptation to dim lighting. She denied nyctalopia or any family history of vision problems. Past ocular and medical histories were unremarkable. She denied drug use. Family history was unrevealing.
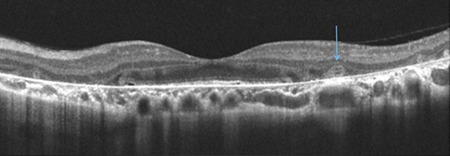
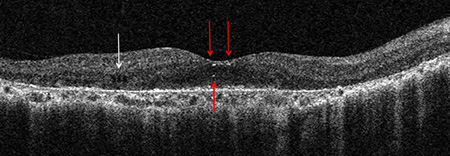
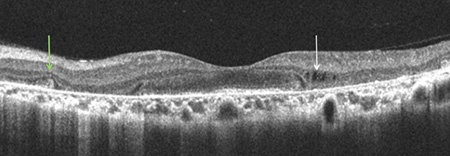
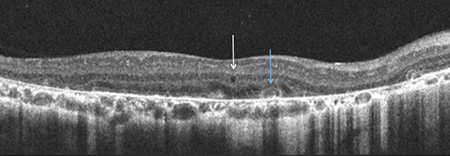
On examination, best visual acuity was 20/25 in the right eye and 20/20 in the left eye. Intraocular pressure and confrontation visual fields were full in both eyes. Anterior segment examination was unremarkable in both eyes and there were no corneal crystals. Posterior segment examination revealed diffuse intraretinal crystalline deposits, with a prominent yellow sheen in the macula bilaterally (Figure 1). There also appeared to be thinning and mottling of the RPE with scattered areas of RPE hyperplasia. There was diffuse, tapetoretinal generation that was prominent in the posterior pole and the mid-periphery (Figure 2). The optic nerve and vessels had a normal appearance. SD-OCT demonstrated diffuse thinning of the outer retina, RPE and choriocapillaris in both eyes (Figure 3). There are also focal areas of inner retinal cystoid spaces and outer retinal tubulation. Infrared reflectance further demonstrated the extensive crystal deposition (Fig 4). Fundus autofluorescence revealed extensive areas of nummular retinal pigment epithelial atrophy (Figure 5).
A
B
D
C
Figure 3: SD-OCT demonstrates gross degeneration/atrophy of the outer retina, RPE and choriocapillaris. Intraretinal crystals (A, red arrows) are seen but have a less discrete appearance when compared to fundus photography. Other findings include inner-retinal cystoid spaces (A,C,D. white arrows), outer retinal tubulation (B,C, blue arrows) and RPE clumping (D, Green Arrow).
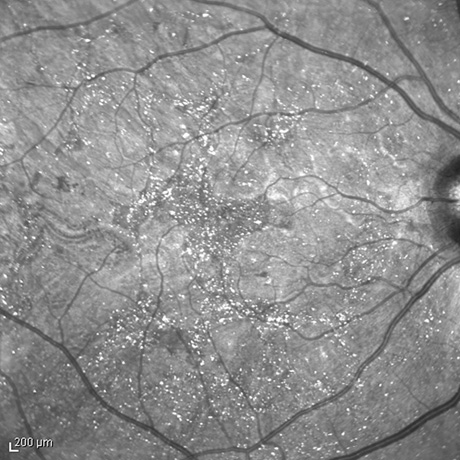
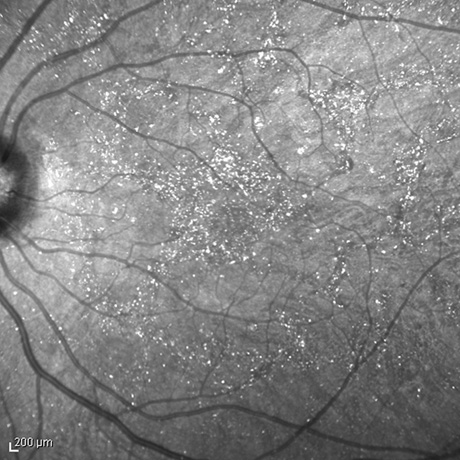
Figure 4: Infrared imaging of the right and left eye highlighting the distribution of the retinal crystals.
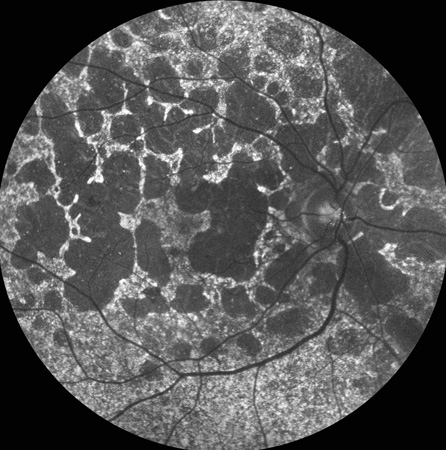
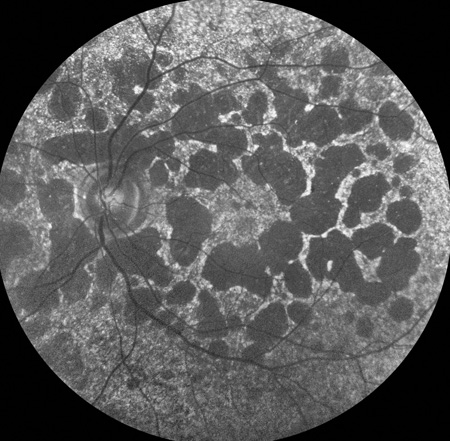
Figure 5: Fundus autoflouresence demonstrates nummular and geography areas of RPE atrophy.
What is your Diagnosis?
Differential Diagnosis
The differential diagnosis for crystalline retinopathies includes; genetic disease such as Bietti’s crystalline retinopathy, gyrate atrophy, cystinosis, Sjogren-Larrson syndrome, primary hereditary hyperoxaluria; metabolic disorders such as secondary hyperoxaluria and diseases associated with hyperabsoption of oxalate including sarcoidosis, liver cirrhosis, small bowel resection and renal failure; vascular disease such as juxtafoveal telangiectasis; and toxic exposures such as canthaxanthine, tamoxifen, nitrofuranatoin, methoxyflurane, and ethylene glycol.
Discussion
Discovered in 1937, Bietti’s crystalline dystrophy is an autosomal recessive tapetoretinal degeneration characterized by intraretinal crystal formation throughout the fundus and perilimbal cornea. Although there have been reports of autosomal dominant inheritance, most reports are likely autosomal recessive. Bietti’s is slowly progressive and is usually diagnosed in adulthood, typically among men. Wilson and colleagues have suggested there maybe two subtypes of disease, one with diffuse involvement leading to profound symptoms and electrophysiologic abnormalities and a more localized form with severe symptoms and a more intact electroretinogram. Although there may be gradual worsening of vision, disease progression is variable and nyctalopia is usually absent. Patients may present in the second decade or even into the sixth decade of life with good Snellen visual acuity. As paracentral scotomas enlarge, tasks such as reading become more difficult. Most reported cases have been among people of Italian or east-Asian decent, especially Japanese and Chinese. Several mutations of the CYP4V2 gene have been associated with this phenotype. This gene encodes a protein in the cytochrome P450 protein superfamily that is responsible for a variety of oxidative metabolic reactions. Although the histopathology of the retinal crystals is unknown, crystalline formation is thought to arise from reduced fatty acid precursor metabolism, and potentially from high intracellular triglycerides and cholesterol levels. Corneal crystals have been reported in one-fourth of patients and are described as fine, white-yellow crystals within the stroma of the perilimbal area. The corneal crystals gradually disappear. Crystals are also located throughout all retinal layers, which is confirmed by spectral domain OCT analysis. Fluoresence angiography demonstrates geographic areas of atrophy of the chorioretinal and choriocapillaris, which become progressively larger over time, revealing the larger vessels of the choroid. The areas between involved and more normal appearing retinal pigment epithelium are well demarcated. Fundus autoflourescence shows geographic areas of hypoautoflouresence consistent with progressive chorioretinal atrophy, punctate areas of hyperautoflourence consistent with hyperpigmentation, and minimal hyper-autoflouresence of the crystals. The optic disc and vessels maintain a normal appearance. Electro-oculogram and electroretinogram typically correlate with the severity of disease. When the extent of involvement is localized or regional, electroretinogram can be normal or moderately abnormal. In more diffuse, widespread Bietti’s, the studies are typically severely abnormal or extinguished. Electronegative electroretinograms have been reported.
Clinical Follow-up
Over 3 years of follow-up, the vision and clinical appearance remained stable. The patient is not interested in pursuing further genetic testing for her condition at this time.
TAKE HOME POINTS:
- Bietti’s crystalline dystrophy is an autosomal recessive disease characterized by retinal crystals, slowly progressive tapetoretinal degeneration and corneal crystals.
- Despite widespread retinal degeneration, patients usually retain good central vision
Want to Subscribe to Case of the Month?
References
- Agarwal A. Gass’ Atlas of Macula Diseases, 5th Ed. Elsevier; 2012. Pg. 346-9.
- Fong AM, Koh A, Lee K, et al. Bietti’s crystalline dystrophy in Asians: clinical, angiographic and electrophysiological characteristics. Int Ophthalmol 2008, Oct 15.
- Lai TY, Ng TK, Tam PO, et al. Genotype analysis of Bietti’s crystalline dystrophy in patients with CYP4V2 mutations. Invest Ophthalmol Vis Sci 2007;48:5212-20.
- Nakano M, Kelly EJ, Rettie AE. Expression and characterization of CYP4V2 as a fatty acid omega-hydroxylase. Drug Metab Dispos 2009;37:2119-22.
- Querques G, Quijano C, Bouzitou-Mfoumou R, et al. In-vivo visualization of retinal crystals in Bietti’s crystalline dystrophy by spectral domain optical coherence tomography. Ophthalmic Surg Lasers Imaging 2010;Mar 9:1-3.
- Wada Y, Abe T, Shioo T, et al. Specular microscopic findings of corneal deposits in patients with Bietti’s crystalline corneal retinal dystrophy. Br J Ophthalmol 1999;83:1095.
- Wilson DJ, Weleber RG, Kelin ML, et al. Bietti’s crystalline dystrophy. A clinicopatholgic correlative study. Arch Ophthalmol 1989;107:213-21.