West Coast Retina
Case of the Month
July, 2011
A 31-year-old male with poor night vision.
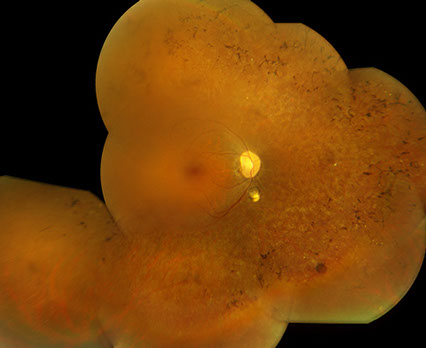
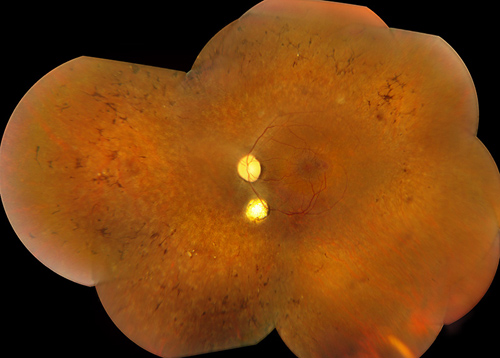
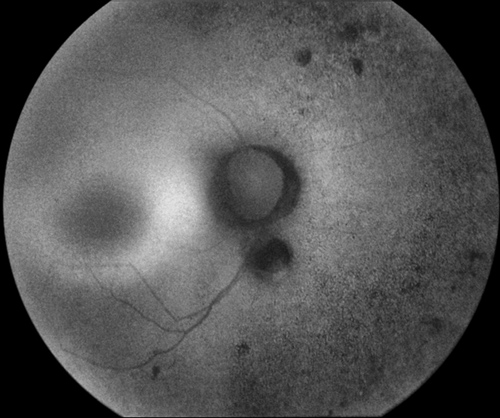
Figures 1: Color fundus photograph montages of the both eyes reveal waxy pallor of the optic nerves, severe arteriolar attenuation, marked peripheral bone spicule pigmentary changes, and a coloboma inferior to the optic nerve.
Case History
A 31-year-old male presented with poor night vision for three months. His past medical history was significant for obesity, hypopituitarism, diabetes mellitus and developmental delay since childhood. He had been on supplemental growth hormone as a child, and was presently taking oral metformin, glyburide, and rosiglitazone for diabetes. He was part of a six sibship family with no history of inherited ocular disease.
On examination, visual acuity was 20/80 in the right eye and 20/50 in the left eye. Intraocular pressures were normal and there was no relative afferent pupillary defect. Confrontation visual field revealed severe constriction with a central island remaining.
Slit lamp examination revealed mild posterior subcapsular cataract in both eyes. Dilated fundus examination revealed waxy pallor of the optic nerves, severe arteriolar attenuation and marked peripheral bone spicule pigmentary changes. In addition, a well-circumscribed area of chorioretinal atrophy (coloboma) was seen inferior to the optic disc, both eyes (Figure 1).
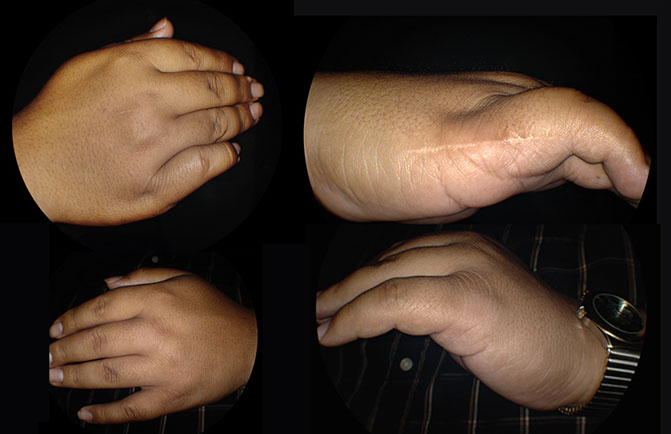
General physical examination was significant for an incisional scar along the medial aspect of the right hand (Figure 2). The patient subsequently confirmed the scar corresponded to surgical removal of an extra digit.
Figure 2: Photographs of both hands show short, stubby fingers, and a surgical scar along the medial aspect of the right hand.
What is your Diagnosis?
Differential Diagnosis
The differential diagnosis of syndromic pigmentary retinopathy includes Alagille syndrome, Alstrom disease, Bassen-Kornzweig syndrome, Cockayne syndrome, Flynn-Aird syndrome, Hallervorden-Spatz syndrome, Zellweger syndrome, Usher syndrome, Olivopontocerebellar dystrophy, Friedrich ataxia, spinocerebellar ataxia type-7, mucopolysaccharidosis, neonatal adrenoleukodytrophy, neuronal ceroid lipofuscinosis (Batten disease), Senior-Loken syndrome, Refsum disease and Bardet-Biedl syndrome.
Clinical Course
The electroretinogram revealed undetectable responses on combined response, rod response, and cone response (with an undetectable peak or trough on the 30Hz flicker) in both eyes, indicating bilateral severe generalized rod and cone dysfunction (Figure 3). The electrooculogram revealed an Arden ratio (light peak to dark trough) of 1.2 in the right eye and 1.1 in the left (normal 1.85), consistent with severe phototransduction abnormalities at the retinal pigment epithelial interface (Figure 4).
Fundus autofluorescence revealed a perifoveal ring of hyper-autofluorescence (Figure 5, characteristic of rod-cone dystrophy).1 Spectral domain optical coherence tomography demonstrated the absence of the photoreceptor inner segment-outer segment (IS/OS) junction and thinning of the outer nuclear layer (with a decrease in thickness centrifugally) also consistent with rod-cone dystrophy (Figure 6). 1
A rod-cone dystrophy in the setting of developmental delay, hypopituitarism, diabetes, obesity and polydactyly pointed to the diagnosis of Bardet-Biedl syndrome. Genetic testing was performed which confirmed the diagnosis. Our patient was evaluated by his primary care physician for the other manifestations of the syndrome. Subsequently, he enrolled in a Bardet-Biedl program sponsored by the National Institutes of Health for ongoing counseling, genetic study, and research-based treatment.
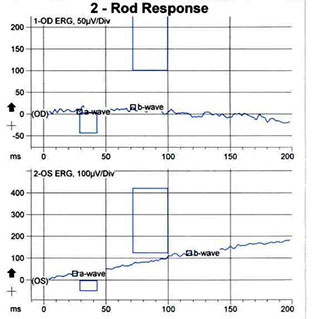
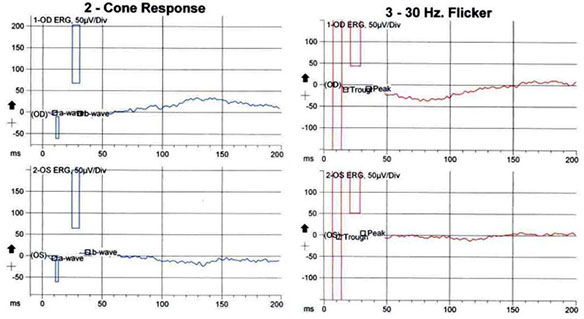
Figures 3: The electroretinogram revealing an undetectable a- and b-wave on the maximal combined response, rod response, and cone response (with an undetectable peak or trough on the 30Hz flicker), indicating severe generalized rod and cone dysfunction.
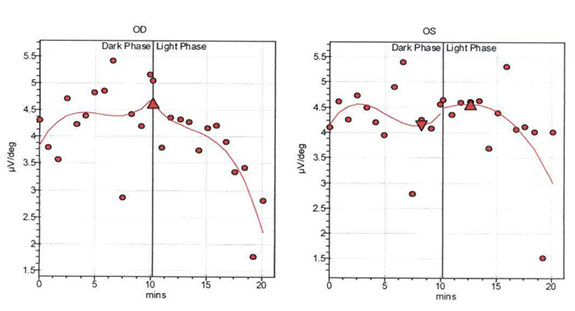
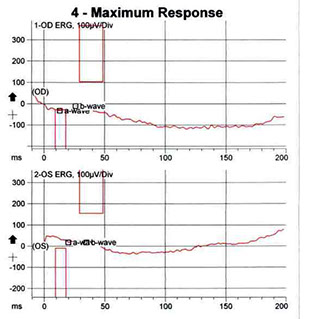
Figure 4: The electrooculogram revealing an Arden ratio (light peak to dark trough) of 1.2 in the right eye and 1.1 in the left (normal 1.85), indicating severe phototransduction abnormalities at the RPE interface.
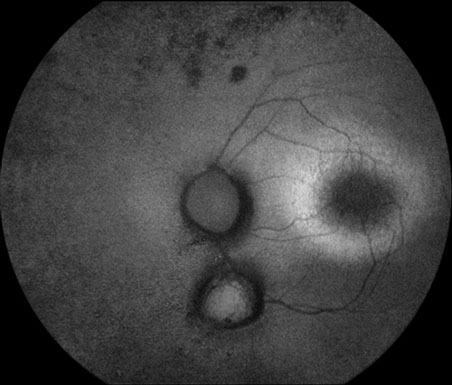
Figures 5: Fundus autofluorescence revealing a perifoveal ring of hyperfluorescence characteristic of rod-cone dystrophy.
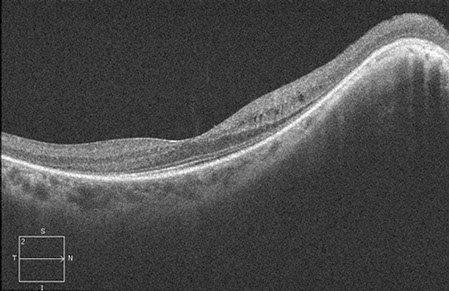
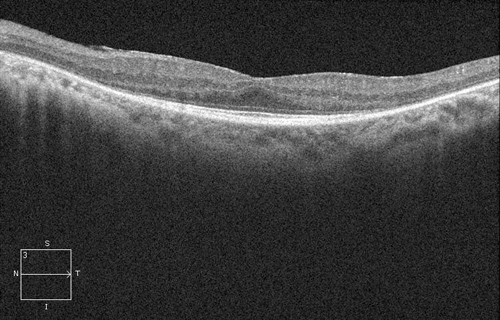
Figures 6: Spectral domain OCT demonstrated the absence of the IS/OS junction and thinning of the outer nuclear layer (with a decrease in thickness centrifugally) consistent with rod-cone dystrophy.
Discussion
Bardet-Biedl syndrome is a genetic syndrome, first described by Bardet in 1920 and Biedl in 1922.2-3 It consists of the following 5 cardinal features:
1. pigmentary retinopathy (92% to 100%)
2. obesity (92% to 96%)
3. postaxial polydactyly (58% to 74%)
4. mental retardation (41% to 87%)
5. hypogonadism in males (74% to 96%).4,5
More recently, renal disease has been suggested as a sixth cardinal feature. The hands often appear puffy with indistinct knuckles. Several other abnormalities that occur less frequently, such as diabetes mellitus and dental disease, are also associated with this syndrome.4 Based on features of the disease, Beales and colleagues proposed diagnostic criteria for this syndrome (Table 1). 4
Bardet-Biedl syndrome was found to occur at a frequency of approximately 1:160,000 in a study carried out on a Swiss population. It is the second most common cause of syndromic retinitis pigmentosa after Usher syndrome. The wide clinical spectrum observed in Bardet-Biedl syndrome is associated with significant genetic heterogeneity. It is now known that additional genes are involved in modification of the expression of the primary gene; hence, the inheritance is now designated as oligogenic rather than simple autosomal recessive. To date, mutations in 12 different genes (BBS1 to BBS12) have been identified as being responsible for this phenotype.6 Although we have expanding knowledge of the particular genes involved in the disease, the diagnosis is presently still made on a clinical basis.
The pigmentary retinopathy associated with Bardet-Biedl syndrome is usually an atypical rod-cone dystrophy with relatively early macular involvement. It typically becomes symptomatic in the first decade of life, with substantial visual loss by the third decade. The electroretinogram is often normal up to the age of 5 years, but by age 10 is significantly attenuated. Other reported ocular features include myopia, cataracts, ptosis, microphthalmia, nystagmus, glaucoma, and strabismus.7-9 Of interest, coloboma as seen in our patient is not described as a characteristic feature in Bardet-Biedl syndrome.
Management of a patient as recommended by Beales and colleagues include the following baseline studies: electroretinogram/visually evoked responses, a renal ultrasound, intravenous pyelogram, echocardiogram, and also recommended consideration of CT or MRI scans of the brain and kidneys, ensuring the patient's educational needs are addressed, registration of blindness, and speech assessment and therapy. Routine health surveillance includes urinalysis once every 6 months, and blood pressure, blood urea nitrogen, and creatinine level assessments once a year.4
Table 1
Diagnostic Criteria for Bardet-Biedl Syndrome4
Primary Features
Rod-cone dystrophy
Polydactyly
Obesity
Learning disabilities
Hypogonadism in males
Renal abnormalities
Secondary Features
Speech disorder/delay
Other ocular abnormalities: strabismus/cataract/astigmatism
Brachydactyly/syndactyly
Developmental delay
Polyuria/polydipsia (nephrogenic diabetes insipidus)
Ataxia/poor coordination/imbalance
Mild spasticity (especially lower limbs)
Diabetes mellitus
Dental crowding/hypodontia/small roots/high arched palate
Congenital heart disease
Hepatic fibrosis
For diagnosis, 4 primary features, or 3 primary and 2 secondary features must be present.
Want to Subscribe to Case of the Month?
Take Home Points
- Patients with a rod-cone dystrophy secondary to Bardet-Biedl syndrome can be easily recognized by obesity and the presence or history of polydactyly. It may be necessary to inspect hands and feet for signs of scars from infantile excision of extra digits.
- These patients should be managed together with their primary care physicians given the wide spectrum of possible systemic anomalies.
- These patients tend to have more severe retinopathy and learning disabilities and extra effort should be made to obtain special educational support.
References
- Lima LH, Cella W, Greenstein VC, Wang NK, Busuioc M, Smith RT, Yannuzzi LA, Tsang SH. Structural assessment of hyperautofluorescent ring in patients with retinitis pigmentosa. Retina. 2009 Jul-Aug;29(7):1025-31.
- Bardet G. Sur un syndrome d'obýsitý congýnitale avec polydactylie et rýtinite pigmentaire (Contributon a l'ýtude des formes cliniques de lýbýsitý hypophysaire). These de Paris. 1920;470:9-107.
- Biedl A. Ein Geschwisterpaar mit adiposo-genitaler Dystrofie. Deutsch Med Wochenschr. 1922;4:1630.
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36:437-446.
- Green JS, Parfrey PS, Harnett JD, et al. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med. 1989;321:1002-1009.
- Sheffield VC, Nishimura D, Stone EM. The molecular genetics of Bardet-Biedl syndrome. Opin Genet Dev. 2001 Jun;11(3):317-21.
- Green JS, Parfrey PS, Harnett JD, et al. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med. 1989;321:1002-1009
- Rizzo JF 3rd, Berson EL, Lessell S. Retinal and neurologic findings in the Laurence-Moon-Bardet-Biedl phenotype. Ophthalmology. 1986;93:1452-1456.
- Schachat AP, Maumenee IH. Bardet-Biedl syndrome and related disorders. Arch Ophthalmol. 1982;100:285-288.