West Coast Retina
Case of the Month
July, 2012
Presented by Paul Stewart, MD
A 42-year-old woman with photopsias and a scotoma in her left eye.

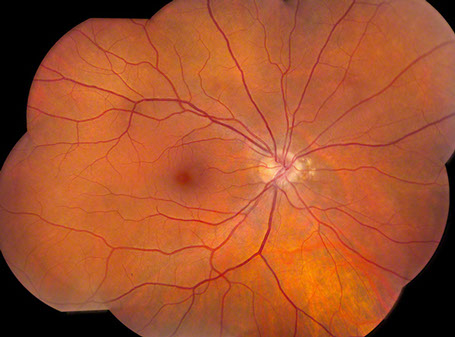
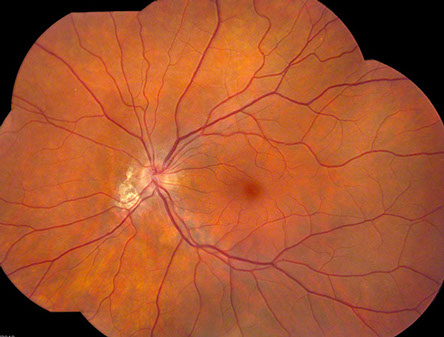
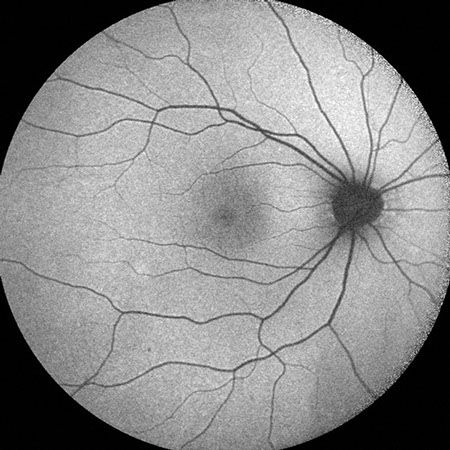
Figures 1 and 2: Color fundus photograph montage of right and left eye. Note the nasal peripapillary pigment atrophy in both eyes.
Case History
A 42 year-old woman presented with a five month history of shimmering photopsias in the left eye associated with a “dark spot” temporal to fixation. Her visual acuity remained unchanged. She was in good health and had no significant past medical or ocular history other than myopia.
On examination her visual acuity was 20/16 in the right eye and 20/13 in the left eye. Intraocular pressure and anterior segment examinations were normal in both eyes. Visual fields were full to confrontation in both eyes. The dilated funduscopic examination of the right eye revealed an area of pigment atrophy and clumping just nasal to the nerve and was otherwise unremarkable (Figure 1). The dilated funduscopic examination of the left eye revealed a more extensive area of atrophy and pigment clumping nasal to the optic nerve (Figure 2).
Fluorescein angiography of the right eye showed normal vascular filling without evidence of window defect or edema (Figure 3). Fluorescein angiography of the left eye showed normal retinal vascular filling (Figure 4). There is an area of hyperfluorescence and hypofluorescence nasal to the optic disc, corresponding to the area of pigment atrophy and clumping.
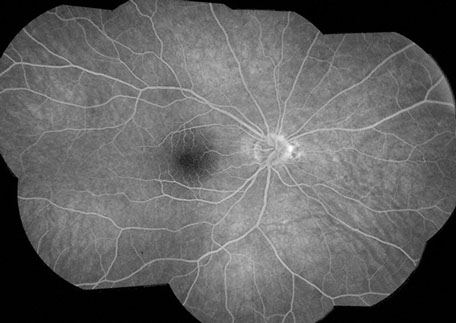
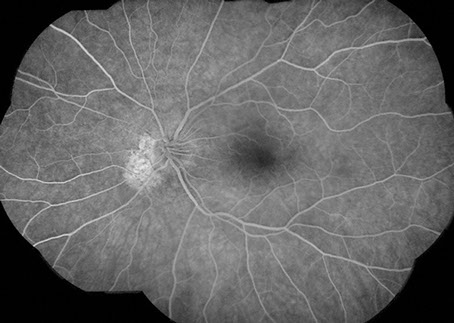
Figures 3 and 4: Fluorescein angiogram montage of the right and left eye. Note the nasal peripapillary areas of hyperfluorescence in each eye though more extensive in the left eye than the right. The study is otherwise unremarkable.
What is your Diagnosis?
Differential diagnosis
The differential diagnosis in this 42 year-old woman with shimmering photopsias and a paracentral scotoma in the left eye includes acute zonal occult outer retinopathy (AZOOR) and an autoimmune retinopathy (AIR), such as cancer associated retinopathy (CAR), melanoma associated retinopathy (MAR), or non-neoplastic autoimmune retinopathy (npAIR). The differential diagnosis also includes various members of the white dot syndromes, such as multiple evanescent white dot syndrome (MEWDS), acute idiopathic enlarged blind spot syndrome (AIBSE), punctate inner choroidopathy (PIC), multifocal choroiditis and panuveitis (MFC), acute macular neuroretinopathy (AMN) and presumed ocular histoplasmosis syndrome (POHS). Consideration should also be given to birdshot chorioretinopathy, acute posterior multifocal placoid pigment epitheliopathy, diffuse unilateral subacute neuroretintitis (DUSN), serpiginous choroidopathy, and idiopathic acute optic neuritis. If there were vitreous cells or other signs of inflammation present the differential could be expanded to include sarcoidosis, Behcet’s, rheumatoid vasculitis and idiopathic retinal vasculitis.
Additional Investigations:
SD-OCT of the right eye showed normal foveal contour and normal retinal architecture (Figure 5). SD-OCT of the left eye showed loss of the IS/OS junction and outer nuclear layer in the peripapillary region (red arrows, Figure 6).
Fundus autofluorescence of the right eye was unremarkable (Figure 7). Fundus autofluorescence of the left eye showed an area of hyperautofluorescence around the optic nerve with a hyperautofluorescent border (Figure 8).
Electroretinography was performed that showed a mild decrease in the A-wave amplitude of the left compared to right eye. There was also a mildly delayed implicit time with mildly reduced amplitude in the left eye. These findings were thought to be consistent with some very mild photoreceptor differences in the left eye (Figure 8).
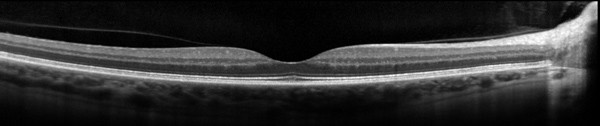
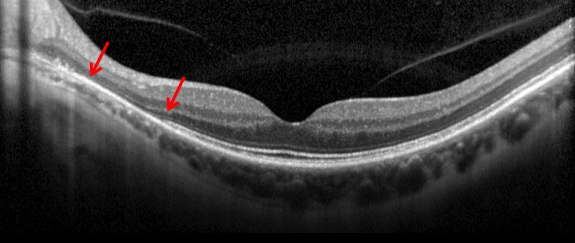
Figures 5 and 6: SD-OCT of the right and left eye. The study in the right eye is normal. In the left eye, note the loss of outer retinal layers, with loss of IS/OS junction (ellipsoid layer) and outer nuclear layer (red arrows and inset).
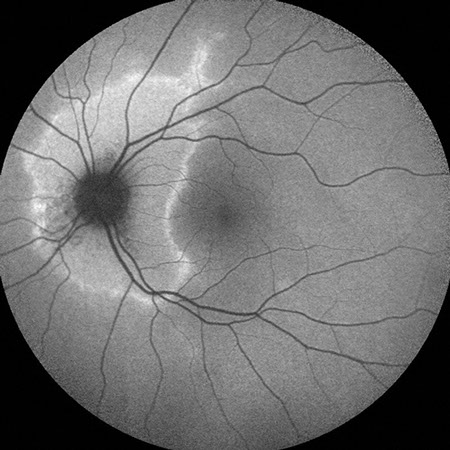
Figures 7 and 8: Fundus autofluorescence of the right and left eye. The right eye is normal. The left eye demonstrates a large circumpapillary area of hyperautofluorescence with a margin of even greater hyperautofluorescence.
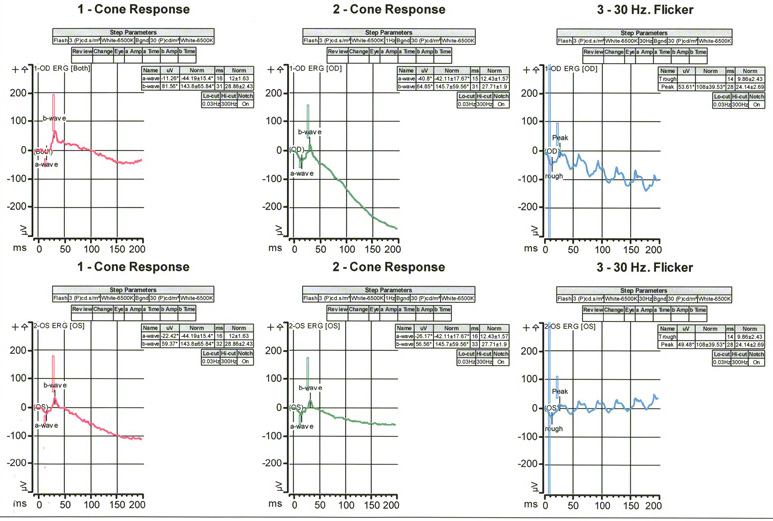
Figure 9: Electrophysiology study of the right and left eye. There is a mild decrease in the A-wave amplitude of the left eye compared with the right eye. Also seen is a mild delay in the implicit time and reduced amplitudes in the left eye.
Clinical Course
The patient has been followed for a year with stable exam and imaging findings. She continues to have a persistent scotoma. Based on her symptoms, ERG and imaging findings she was diagnosed with AZOOR.
Discussion
Acute zonal occult outer retinopathy was first described by Gass in 1992 with a report of 13 patients, mostly young women, with acute loss of zones of outer retinal function, photopsia, ERG changes and minimal funduscopic changes.1 Since the first reports there have been more than 130 patients described in the literature.2 Some patients have a viral prodrome. Occasionally vitreous inflammation develops, notably in those with a large zone of visual field loss.3 The visual field usually stabilizes in 4-6 months and sometimes will improve. Although unilateral in presentation more than 60 percent of the time, there can be delayed involvement of the fellow eye. There can also be recurrences in the same eye.
A variant of AZOOR, known as acute annular outer retinopathy (AAOR) was first described in 1994 by Luckie and Ai, et al, in which patients have an acute onset of scotoma.4 This entity reveals a gray intraretinal ring at the edge of the scotoma.
Gass proposed the pathogenesis to be related to an infection of the retinal photoreceptors adjacent to the optic nerve, likely from a viral source.5 He felt that the white dot syndromes were related entities that typically occur in young women with a similar constellation of findings. Jampol and Becker have presented an alternative theory with a common genetic hypothesis of autoimmune/inflammatory diseases.6 They hypothesize that there is an interplay of genetics, the immune system and environmental triggers that lead to specific diseases such as AZOOR.
There has been no treatment proven to improve outcomes in AZOOR. There are several reports of attempted corticosteroid use. Gass felt that oral or intravenous steroids made no difference in outcomes.3 A recent report documents improved visual function in a patient with steroid pulse therapy.7
There have been several reports recently looking at spectral domain OCT imaging as well as autofluorescence. Just as in our case, there is disruption of the IS/OS junction in the region of the scotoma and occasionally loss of the outer nuclear layer.8 Abnormalities of RPE function as shown by autofluorescence have also been reported.9 While most patients in Spaide’s paper demonstrated hypoautofluorescence, there were some focal areas of hyperautofluorescence. Cohen and Jampol report a case with a hyperfluorescent border on autofluoresence, as we also see in our case.10 The hyperautofluorescence seen in our patient may be due to an earlier finding in the evolution of this disorder. It will be interesting to see if this finding evolves and shows hypoautofluorescence with longer follow-up.
Fluorescein angiography was noted to be abnormal in only 9% of patients in Gass’ series.3 These FA exams were done at presentation. Other cases have shown angiographic changes in about 50% of patients, primarily related to changes in the RPE.2 Our patient showed only minimal changes with very mild hyperfluoresence in the peripapillary region of the left eye, corresponding to an area of RPE atrophy. ICG has been reported in 8 patients and primarily showed hypfluoresence in the area of visual field defect.2
Electroretinography is abnormal in the majority of AZOOR cases. In Gass’ long-term study every patient had an abnormal ERG.3 Electo-oculograms (EOG) have also been performed on 39 patients in the literature and show a reduction in the light rise in every case tested.2
Recently Mkrtchyan, Lujan et al reported on the outer retinal structure of AZOOR patients using adaptive optics scanning laser ophthalmoscopy (AOSLO).11 AOSLO can be used to generate high-resolution en-face images of the retina, which can show cone spacing in various locations. This group was able to demonstrate regions of cone loss, alteration in cone structure, as well as areas of normal cone mosaic in the region of visual loss. This paper was the first report of in vivo cone spacing and rod imaging in AZOOR patients.
In summary, our patient has AZOOR with the classic symptoms of photopsias and visual field loss. On imaging we demonstrate some of the characteristic findings on SD-OCT and autofluorescence. ERG in our case shows mild change in the left eye consistent with her diagnosis.
Take Home Points
- AZOOR is a syndrome with one or more zones of visual dysfunction, usually with photopsias, reduced outer retinal function on ERG, and with occasional photoreceptor cell loss that occurs in young women .
- The pathogenesis is unknown at this point with hypotheses supporting causation from a viral infection and from a specific autoimmune process.
- There is no clear evidence for a treatment to improve visual function, although corticosteroids have been tried with occasional success.
- Advances in imaging with SD-OCT and now with adaptive optics scanning laser ophthalmoscopy enable us to see more of the pathology, where previously the ophthalmoscopic examination was unremarkable in these patients.
Want to Subscribe to Case of the Month?
References
- Gass JD, Acute zonal occult outer retinopathy. Donders Lecture: The Netherlands Ophthalmological Society, Maastricht, Holland, June 19, 1992. J Clin Neuroophthalmol. 1993; 13: 79-97.
- Monson DM, Smith JR. Acute zonal occult outer retinopathy. Surv Ophthalmol. 2011; 56: 23-35.
- Gass JD, Agarwal A, Scott IU. Acute zonal occult outer retinopathy: a long-term follow-up study. Am J Ophthalmol. 2002; 134: 329-339.
- Luckie A, Ai E, Del Piero E. Progressive zonal outer retinitis. Am J Ophthalmol. 1994; 118: 583-588.
- Gass JD. Are acute zonal occult outer retinopathy and the white spot syndromes (AZOOR complex) specific autoimmune diseases? Am J Ophthalmol. 2003; 135: 380-381.
- Jampol LM, Becker KG. White spot syndromes of the retina: a hypothesis based on the common genetic hypothesis of autoimmune/inflammatory disease. Am J Ophthalmol. 2003; 135: 379-379.
- Kitakawa T, Hayasi T, Takashina H, et al. Improvement of central visual function following steroid pulse therapy in acute zonal occult outer retinopathy. Doc Ophthalmol. 2012; 124: 249-254.
- Spaide RF, Koizumi H, Freund KB. Photoreceptor outer segment abnormalities as a cause of blind spot enlargement in acute zonal occult outer retinopathy-complex diseases. Am J Ophthalmol. 2008; 146: 111-120.
- Fujiwara T, Imamura Y, Giovinazzo VJ, Spaide RF. Fundus autofluorescence and optical coherence tomographic findings in acute zonal occult outer retinopathy. Retina. 2010; 30: 1206-1216.
- Cohen SY, Jampol LM. Choroidal neovascularization in peripapillary acute zonal occult outer retinopathy. Retinal Cases and Brief Reports. 2007; 1: 220-227.
- Mkrtchyan M, Lujan BJ, Merino D, Thirkill CE, Roorda A, Duncan JL. Outer retinal structure in patients with acute zonal occult outer retinopathy. Am J Ophthalmol. 2012; 153: 757-768.