West Coast Retina
Case of the Month
JULY, 2014
Presented by Steven Williams, MD
A 45 year-old man presents with flashing lights and decreased vision

A
B
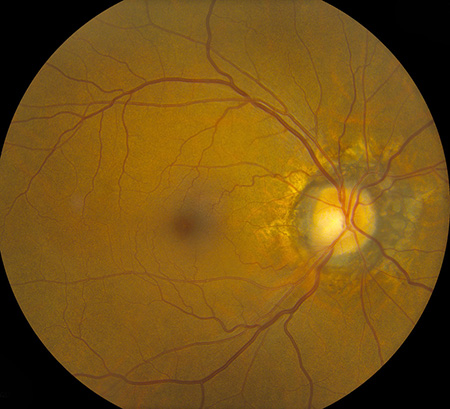
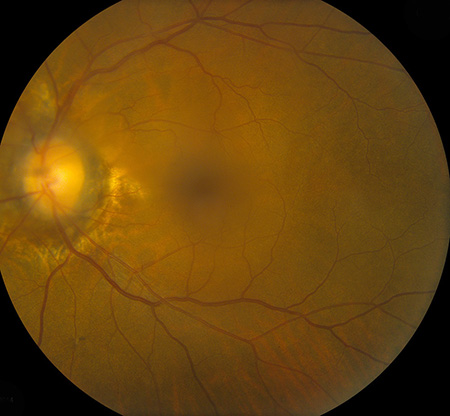
Figures 1A and B: Fundus photograph of the right and left eye. The macula and vessels in the posterior pole appear normal. Note the mild cupping of the optic nerve associated with circumferential periparillary RPE atrophy. Angioid streaks are visible around both nerves. There is vitreous hemorrhage in the left eye.
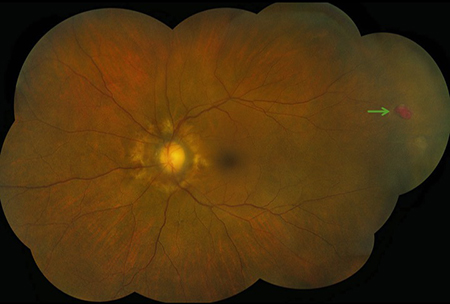
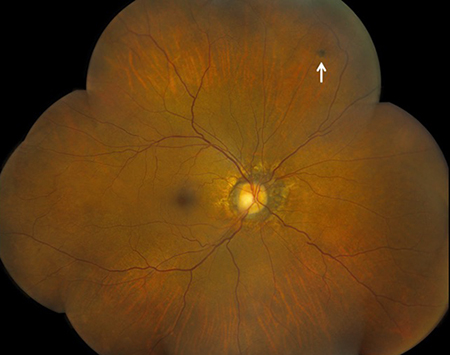
Figures 2A and B: Fundus photograph montage of the right and left eye. Angioid streaks are present, most prominent in the left eye. Note the area of RPE hyperpigmentation superiorly in the right eye (white arrow) and the preretinal hemorrhage in the temporal periphery of the left eye (green arrow, and inset).
B
A

A
B
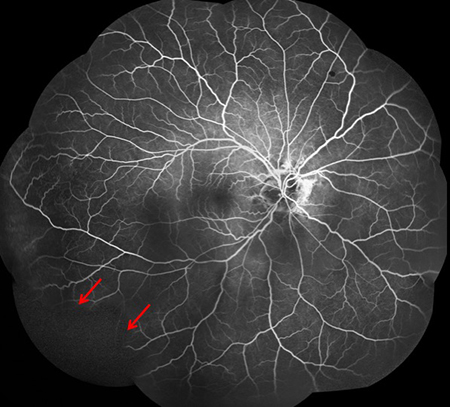
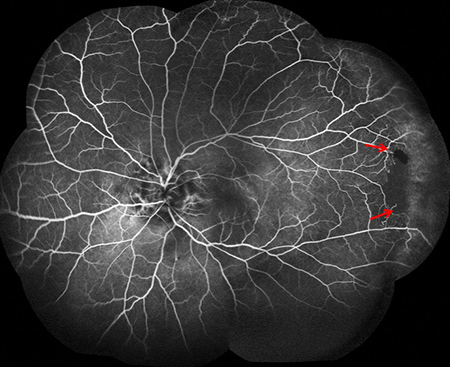
Figures 3A and B: Fluorescein angiogram montages of the right (A) and left (B) fundus. Note the angioid streaks in both eyes. Peripheral nonperfusion is also present in each eye (arrows)
A
B
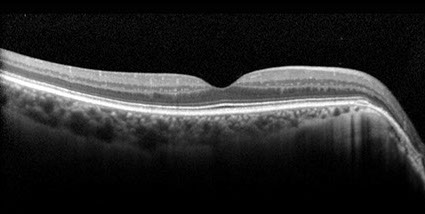
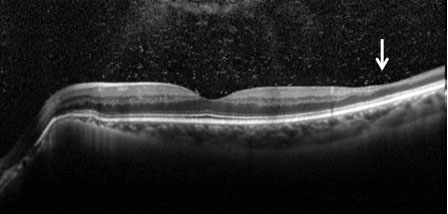
Figures 4A and B: OCT of each macula demonstrated normal foveal contour in both eyes with peripapillary RPE thinning. There was thinning of the inner retina in the temporal macula of the left eye (arrow), most notably of the inner nuclear and inner plexiform layers (B). Vitreous hemorrhage was also observed in the left eye (B).
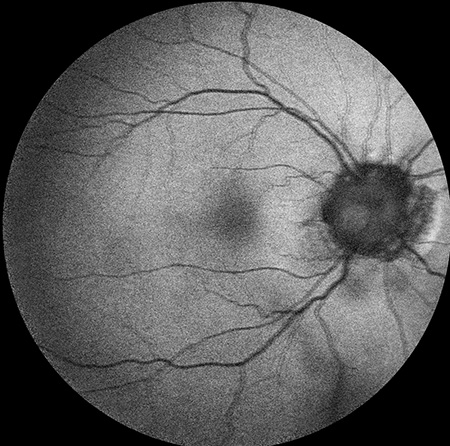
A
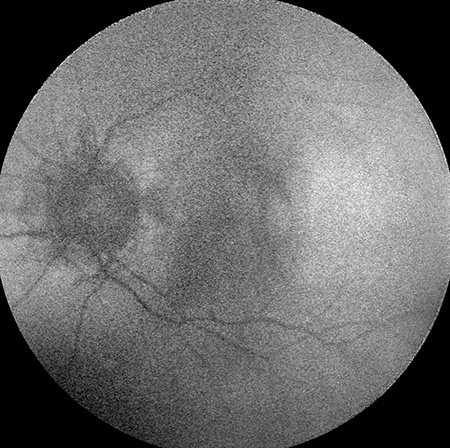
B
Figures 5A and B: Fundus autofluorescence showed subtle presence of angioid streaks and nonspecific peripapillary RPE alterations in both eyes.
What is your Diagnosis?
Diagnosis
The differential diagnosis for peripheral retinal non-perfusion consists of several clinical entities. One major category includes primary retinal vasculopathies, e.g. familial exudative retinal vasculopathy (FEVR), Coats disease, Eales disease. Retinopathies associated with systemic vascular disease include diabetic retinopathy and radiation retinopathy. Inflammatory etiologies known to cause peripheral non-perfusion include multiple sclerosis, pars planitis, systemic lupus erythematous, and sarcoidosis. The final category includes, blood dyscrasias such as sickle-cell retinopathy, polycythemia vera, hemoglobin C trait, and leukemia.
Additional Case History
Our patient had sickle-cell SC disease. He has had numerous hospitalizations for sickle cell crisis and also suffered from hypertension and anemia. He had also undergone a cholecystectomy and was medicated with hydrourea to reduce the occurrence of sickle crisis.
Discussion
Sickle cell retinopathy is commonly seen among people of African descent who have homozygous sickle-cell disease (SS), hemoglobin SC disease (SC), sickle-cell thalassemia (S-thal) and hemoglobin SO Arab. Sickle-cell disease (SS) has autosomal recessive inheritance and is caused by mutations in the beta-globin gene on the short arm of chromosome 11. Hemoglobin SO Arab is a lesser known hemoglobin variant which results from a point mutation on the beta-globin gene. Only 10% of patients with SC and SS disease develop vision loss. Sickle cell retinopathy patients develop circumferential, peripheral occlusive vasculopathy and retinal neovascularization. This leads to intraretinal and subretinal hemorrhage, which in the acute or subacute phase is commonly referred to as a salmon-patch hemorrhage and progresses to an iridescent patch of hemosiderin or hyperplastic RPE called a sunburst spot in the chronic phase. Patients also develop areas of retinal neovascularization referred to as sea fans. Sickle cell retinopathy is also associated with vitreous hemorrhage, central retinal artery (CRAO), central retinal vein occlusion (CRVO), macular hole formation, tractional and rhegmatogenous retinal detachment, ischemic optic neuropathy, choroidal infarction, and neovascular glaucoma. Angioid streaks occur in as many as 22% of patients with SS disease. Most patients develop peripheral vaso-occlusive disease and enlargement of the foveal avascular zone without any symptoms. Goldbaum has also described a characteristic pattern of temporal retinal thinning which is consistent with the thinning of the inner retina seen on OCT in our patient (Figure 4B).
The diagnosis of sickle cell disease is made by hemoglobin electrophoresis. Although SS disease is associated more commonly with systemic symptoms, SC disease is more likely to result in ocular complications. Patients with sickle cell trait rarely have retinal vascular findings. Despite the presence of peripheral ischemia and retinal neovascularization, most eyes do not develop sequelae. Eyes that go on to develop vitreous hemorrhage or more serious sequelae of proliferative retinopathy should be treated. Treatment for retinal neovascularization usually consists of laser photocoagulation or cryotherapy that is applied to the areas of neovascularization as well as to areas of retinal nonperfusion. Acute treatment of CRAO may include the initiation of oxygen therapy to improve oxygen carrying capacity or blood transfusion.
Take Home Points
- Sickle cell retinopathy should be suspected in patients with occlusive peripheral vasculopathy, especially if they are of African descent.
- Diagnosis of sickle cell hemoglobinopathy can be confirmed using hemoglobin electrophoresis.
- Sickle cell retinopathy usually does not require treatment unless sequelae of proliferative disease occur.
Want to Subscribe to Case of the Month?
References
- Clarkson JG. The ocular manifestations of sickle-cell disease: a prevalence and natural history study. Trans Am Ophthalmol Soc 1992;90:481-504.
- Dizon RV, Jampol LM, Goldberg MF, et al. Choroidal occlusive disease in sickle cell hemoglobinopathies. Surv Ophthalmol 1979;23:297-306.
- Downes SM, Hambleton IR, Chuange EL, et al. Incidence and natural history of proliferative sick cell retinopathy: Observations from a cohort study. Ophthalmology 2005;112:1869-75.
- Goldbaum MH. Retinal depression sign indicating a small retinal infarct. Am J Ophthalmol 1978;86:45-55.
- Nagpal KC, Asdourian G, Goldbaum M. Angioid streaks and sickle haemoglobinopathies. Br J Ophthalmol 1976;60:31-4.
- Sanders RJ, Brown GC, Rosenstein RB, et al. Foveal avascular zone diameter and sickle cell disease. Arch Ophthalmol 1991;109:812-5.