West Coast Retina
Case of the Month
June, 2012
A 30-year-old woman with reduced vision in her right eye.
Presented by Sandeep Randhawa, MD
Photography by Ollie Brock
and Chad Indermill

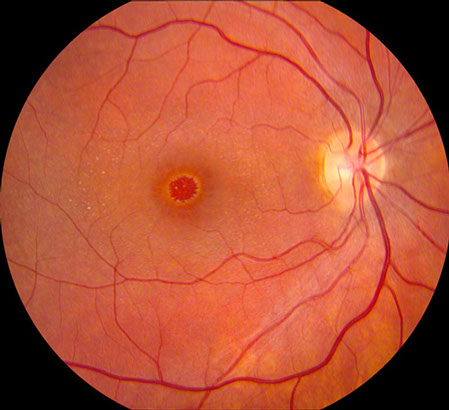
A
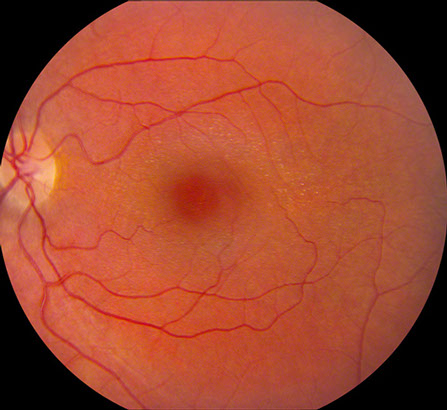
B

C
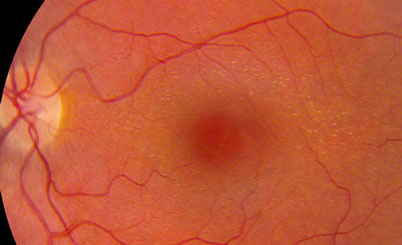
D
Figures 1A-D: Color fundus photographs of the right and left eye. Note the macular hole in the right eye (A and C) and the presence of perimacular flecks in both eyes.
Case History
A thirty-year-old woman presented with decreased central vision in her right eye. She had been noticing distortion in shapes with her right eye for a month. Her past medical history was significant for Alport syndrome which had been diagnosed when she was six years old. She had a renal transplant at age twenty-five for end stage renal disease secondary to glomerulonephritis from Alport syndrome. She was using cochlear implants for her sensorineural hearing loss (also part of her Alport syndrome). She was on tacrolimus and mycophenolate for post transplant immunosupression.
On examination her visual acuity was 20/100 in her right eye and 20/25 in the left eye. Intraocular pressure was normal both eyes. Slit lamp examination of the anterior segment revealed mild anterior lenticonus in both eyes. A dilated fundus examination was performed which showed a macular hole in the right eye and the presence of perimacular flecks in both eyes (Figure 1A and B). The peripheral fundus was unremarkable both eyes. There was no posterior vitreous detachment in either eye.
What is your Diagnosis?
Is there a relationship between Alport Syndrome and the presence of macular hole?
Additional Investigations
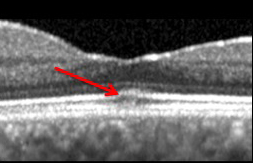
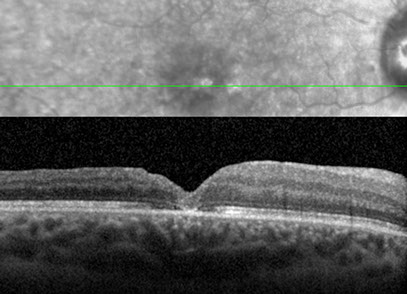
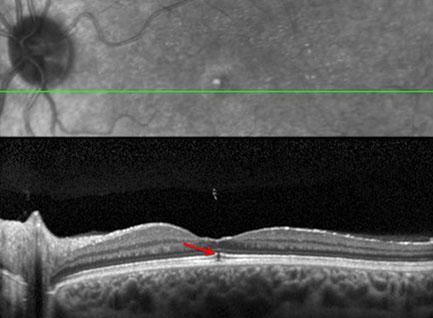
Spectral domain optic coherence tomography (SD-OCT) confirmed the presence of a full thickness macular hole in the right eye with thinning of the temporal macula (Figure 2A). No detachment of the posterior hyaloid was evident. The temporal macular thinning was also evident in the SD-OCT of the left eye. The thinning predominantly involved the inner nuclear layer and the ganglion cell layer. In addition, on the SD-OCT of the left eye, a small, highly reflective region was seen between the inner segment/outer segment (IS/OS) junction and cone outer segment tip (COST) lines at the foveal center. This has been decribed as the ‘cotton ball sign’ after its appearance and its presence is thought to indicate an inward traction on the fovea. (Figure 2B).1
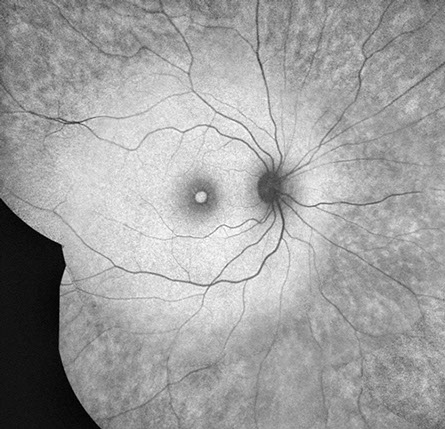
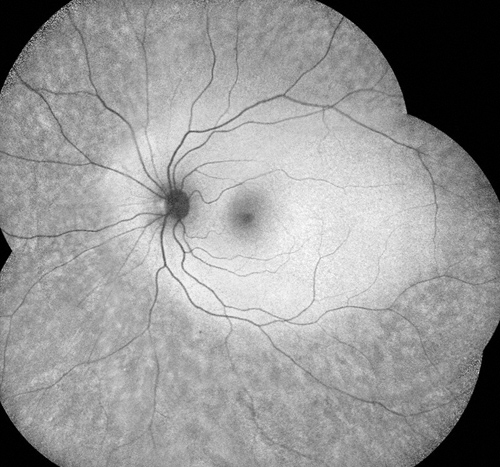
Fundus autofluorescence showed an interesting pattern of splotchy hypoautofluorescence beyond the arcades (in the mid and far periphery both eyes) even though the peripheral exam was normal (Figure 3).
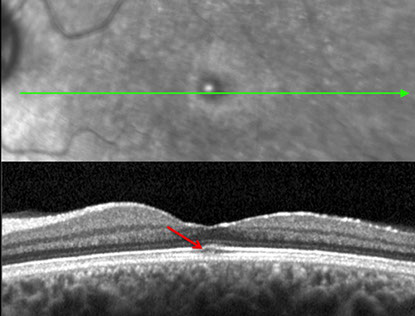
B
A
Figure 2A: SD-OCT of the right macula showing the presence of a full thickness macular hole in the right eye with thinning of the temporal macula.
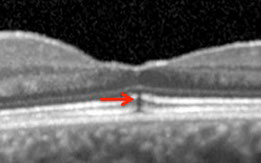
Figure 2B: SD-OCT of the left macula revealing temporal macular thinning predominantly involving the inner nuclear layer and the ganglion cell layer. The arrows point to the highly reflective region seen between the inner segment/outer segment (IS/OS) junction and cone outer segment tip (COST) lines (‘cotton ball sign’) thought to represent inward traction on the fovea. The detailed view shows this more clearly.
A
B
Figure 3A and B: Fundus autofluorescence showing an interesting pattern of splotchy hypoautofluorescence beyond the arcades (in the mid and far periphery both eyes) even though the peripheral exam was normal in both eyes.
Clinical Course
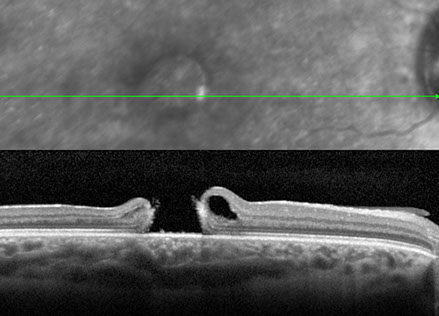
The patient underwent a 25-gauge pars plana vitrectomy, internal limiting membrane (ILM) peeling and fluid gas exchange in the right eye. At the time of the surgery, the posterior hyaloid was found to be very adherent and the hyaloid was detached from the disc using an extrusion needle with high aspiration and then carefully peeled till beyond the vascular arcades using forceps. This was followed by removal of the ILM aided by dilute triamcinolone staining. On the one month post operative visit, the hole was confirmed to be closed on SD- OCT (Figure 4A) and vision had improved to 20/60 in the right eye. Interesting the SD- OCT of the left eye at that visit revealed a focal subfoveal defect at the level of the retinal pigment epithelium (RPE) and the IS/OS junction and separation of the posterior hyaloid from the macula (Figure 4B, as a result of vitreomacular traction sometimes seen as a ‘foveal red spot sign’ on exam and thought to represent a healed macular microhole)2 The visual acuity in the left eye had slightly deteriorated to 20/30 (compared to 20/25 at the initial visit a month prior). The presence of the ‘foveal cotton ball sign’ initially, and the subsequent evolution to the focal subfoveal defect at the IS/OS and RPE on follow up were evidence of dynamic vitreoretinal traction in the left eye.
A
Figures 4A and B: A: SD- OCT of the right eye at the one month post op visit showing closure of the macular hole after surgery.
B: SD-OCT of the left eye revealed a focal subfoveal defect at the level of the retinal pigment epithelium (RPE) and the inner segment-outer segment (IS/OS) junction (arrow, and inset) and separation of the posterior hyaloid from the macula (as a result of vitreomacular traction)
B
Discussion
Alport syndrome is a very rare inherited disease that occurs in 1 of 5,000 to 50,000 live births and is characterized by renal failure, sensorineural hearing loss, lenticonus, and retinopathy. Up to 85% of families have X-linked inheritance3 with mutations in the COL4A5 gene,4 and most of the others have autosomal recessive disease with mutations in the COL4A3 or -4 genes.5 All mutations result in the loss of the α3(IV)–α5(IV) collagen network from affected basement membranes and the subsequent development of thinning or lamellation.6
The main ocular abnormalities in Alport syndrome are anterior lenticonus, and the central and peripheral retinopathies that occur in at least 50% males and 20% females with X-linked inheritance and also commonly in autosomal recessive disease. Anterior lenticonus results from bulging of the lens through a thinned capsule. The central retinopathy comprises whitish-yellow perimacular dots and flecks that are present from early adolescence and are more common when renal failure, hearing loss, and lenticonus are also present.7 The central and peripheral retinopathies do not affect vision or require treatment and may occur independently.8,9
Abnormal collagen synthesis may account for many findings in Alport syndrome. Its role in macular hole pathogenesis may be to accelerate passage of fluid through a structurally abnormal Bruch’s membrane yielding microcystic cavities10 which may coalesce and rupture, or due to potentiated vitreoretinal traction at the vitreoretinal interface.11
Savige and coworkers identified the presence of the α3(IV)–α5(IV) collagen chains in the normal ILM as well as the RPE basement membrane of Bruch's membrane in normal human retina by immunohistochemical staining. In addition, they identified abnormalities (by OCT segmentation analysis on ten patients with Alport syndrome and electron microscopy of an Alport donor cadaveric eye) to help explain the retinal characteristics of Alport syndrome. They found the hyperreflectivity of the ILM/nerve fiber layer (NFL) on OCT corresponding to the distribution of the dot-and-fleck retinopathy and thinning of the temporal macula. In addition, thinning of the ILM and RPE basement membrane of the Bruch's membrane was also seen suggesting that these maybe primary defects that result in the retinopathy, the thinned temporal macula, and, potentially, the macular hole. The ILM (formed by the footplates of the Muller cells) forms a physicial barrier that protects the retina from toxins and from traction from the vitreous as the eye moves. Bruch’s membrane primarily regulates the passage of nutrients and metabolites between the RPE and the underlying choriocapillaris. A thinned ILM could increase susceptibility to vitreoretinal traction and a thinned Bruch’s membrane could lead to abnormal permeability and cysts.
Alport syndrome–associated macular holes appear to be distinguished from idiopathic macular holes by their tendency for larger size, earlier age at onset, and difficulty of surgical repair. Weakness of basement membrane and an abnormal vitreoretinal interface likely contribute to less frequent surgical success.12
In addition, our patient had a pattern of splotchy hypoautofluorescence in the periphery (in the absence of any peripheral retinopathy on exam). We postulate that this was due to abnormalities in the RPE/ Bruch’s membrane secondary to the Alport syndrome.
Take Home Points
- Ophthalmic findings in Alport syndrome, in addition to, anterior lenticonus and flecked retinopathy, include macular hole.
- Macular holes in Alport syndrome are usually larger, and more difficult to repair. Meticulously performed vitrectomy with separation of the posterior hyaloid and ILM peeling with fluid gas exchange leads to successful closure.
Want to Subscribe to Case of the Month?
References
- Tsunoda K, Watanabe K, Akiyama K et al.Highly reflective foveal region in optical coherence tomography in eyes with vitreomacular traction or epiretinal membrane. Ophthalmology. 2012;119:581-7.
- Johnson MW. Posterior vitreous detachment: Evolution and complications of its early stages. Am J Ophthalmol 2010;149:371-382 e1.
- Feingold J, Bois E, Chompret A, Broyer M, Gubler M-C, Grunfeld J-P. Genetic heterogeneity of Alport syndrome. Kidney Int. 1985;27:672–677.
- Barker DF, Hostikka SL, Zhou J, et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science. 1990;248:1224–1227.
- Mochizuki T, Lemmink HH, Mariyama M, et al. Identification of mutations in the alpha3(IV) and alpha 4(IV) collagen gene in autosomal recessive Alport syndrome. Nat Genet. 1994;8:77–81.
- Yoshioka K, Hino S, Takemura T, et al. Type IV collagen _5 chain: normal distribution and abnormalities in X-linked Alport syndrome revealed by monoclonal antibody. Am J Pathol. 1994;44:986–996.
- Zhang KW, Colville D, Tan R, et al. The use of ocular abnormalities to diagnose X-linked Alport syndrome in children. Pediatr Nephrol. 2008;23(8):1245–1250
- Govan JA . Ocular manifestations of Alport's syndrome: a hereditary disorder of basement membranes? Br J Ophthalmol. 1983;67:493–503
- Shaw EA, Colville D, Wang YY, et al . Characterisation of the peripheral retinopathy in X-linked and autosomal recessive Alport syndrome. Nephrol Dial Transplant. 2007;22:104–108.
- Mete UO, Karaaslan C, Ozbilgin MK, et al. Alport’s syndrome with bilateral macular hole. Acta Ophthalmol Scand 1996;74:77-80
- Shaikh S, Garretson B, Williams G. Vitreoretinal degeneration complicated by retinal detachment in Alport syndrome. Retina 2003;23:119–120.
- Savige J, Liu J, DeBuc DC. Retinal basement membrane abnormalities and the retinopathy of Alport syndrome. Invest Ophthalmol Vis Sci. 2010 Mar;51(3):1621-7.