West Coast Retina
Case of the Month
May, 2010
Presented by Evelyn Fu, MD
A 25-year-old asymptomatic, highly myopic woman underwent evaluation of her peripheral retina.
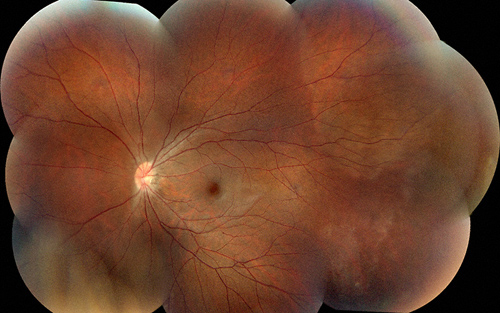
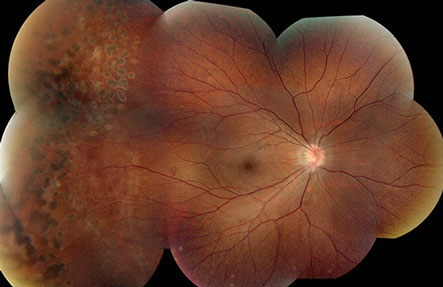
Figures 1A and B: Color montage photographs of the right and left eye show retinal vessel straightening, increased distance between the fovea and optic nerve suggestive of foveal ectopia, as well as prior laser treatment of lattice degeneration and atrophic holes.
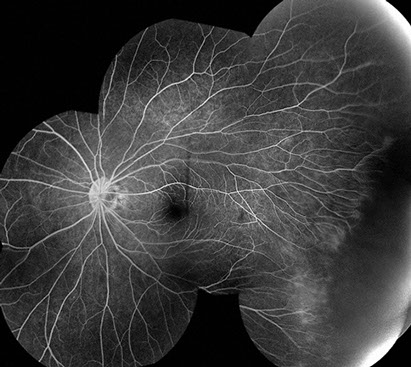
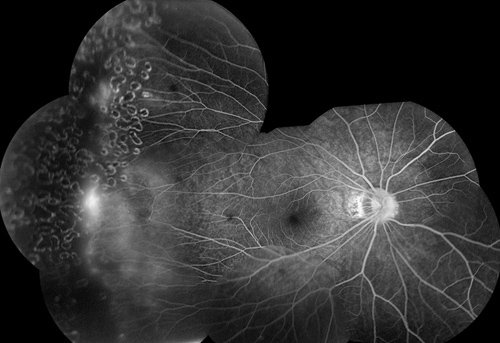
Figures 2A and B: Fluorescein angiography montages of each eye show peripheral areas of telangiectasis, with areas of nonperfusion and neovascularization.
Case History
A 25-year-old asymptomatic woman with high myopia was referred for evaluation of the peripheral retina in both eyes. Her past ocular history is notable for lattice degeneration with atrophic holes treated with laser retinopexy. Her past medical history is non-contributory. There was no history of prematurity or low birth-weight
On examination, visual acuity was 20/20 in the right eye and 20/32 in the left eye. Intraocular pressure and anterior segment examination were normal in each eye. Posterior segment examination of both eyes showed tilted discs with peripapillary atrophy. The retinal vessels were straightened with telangiectasia and neovascularization in the temporal periphery (Figures 1A and B). The distances between the optic nerve and fovea appear increased, suggestive of macular ectopia. Both eyes had treated areas of lattice degeneration with atrophic holes.
Fluorescein angiography of both eyes showed telangiectatic vessels in the temporal periphery with ischemia anterior to these dilated vessels. There was progressive hyperfluorescence due to leakage from the neovascularization seen on clinical examination (Figures 2A and B).
What is your Diagnosis?
Differential Diagnosis
The differential diagnosis of peripheral retinal ischemia and neovascularization is extensive and includes many vascular and inflammatory diseases. Important conditions to consider include: retinopathy of prematurity (ROP), familial exudative vitreoretinopathy (FEVR), embolic retinal vascular disease (eg, talc), hemoglobinopathies, intermediate uveitis, occlusive retinal vasculitis, juvenile retinoschisis and incontinentia pigmenti (IP). The presence of macular ectopia narrows the differential diagnosis to developmental vascular abnormalities including: ROP, FEVR, IP and retinoschisis. Base on our patient’s bilateral clinical findings and a lack of systemic symptoms, she was suspected to have FEVR. Genetic testing was performed and a mutation in frizzled-4 (FZD4), a gene associated with autosomal dominant FEVR, was found.
Overall, the spectrum of findings in FEVR is most similar to ROP. However, unlike patients with ROP, FEVR patients do not have a history of prematurity, low birth-weight, and perinatal complications. Additionally, there are often family members with affected vision or evidence of disease on screening. Regression of vascularization of the peripheral retina is never observed in FEVR but common in ROP.
Incontinentia pigmenti is an X-linked disease that can present with avascular peripheral retina, peripheral neovascularization, and retinal dragging or detachment. Like FEVR, it can manifest in infancy or later in life and can progress rapidly. However, IP is only seen in females as the condition is lethal in males. Most importantly, unlike FEVR, IP is associated with a number of systemic findings: characteristic blistering of the skin in the neonatal period followed by depigmentation of the skin, central nervous system abnormalities, dental hypoplasia, and alopecia.
FEVR may simulate juvenile X-linked retinoschisis when there is peripheral vitreoretinal traction with dragged or detached retina. Findings that are suggestive of juvenile retinoschisis are inferior retinal schisis cavities, characteristic “spoke-wheel” foveal schisis, and a lack of avascular periphery and neovascularization.
Discussion
Familial exudative vitreoretinopathy (FEVR) is a hereditary vitreoretinal dystrophy first described by Criswick and Schepens in 1969.1 The classic and most common mode of inheritance is autosomal dominant with near 100% penetrance. However, X-linked recessive and autosomal recessive inheritance have also been reported. Clinically, it is characterized by incomplete vascularization of the peripheral retina that result from a premature arrest of retinal angiogenesis or retinal vascular differentiation.2 Mild forms of the disease often are asymptomatic with only peripheral vascular abnormalities such as a peripheral avascular zone, vitreoretinal adhesions, arteriovenous anastomoses, numerous vascular branching, and a V-shaped area of retinochoroidal degeneration. More severe manifestations include neovascularization, subretinal and intraretinal hemorrhage and exudates, vitreous hemorrhages, and vitreoretinal traction that can cause retinal folds, macula ectopia, and retinal detachment. The clinical manifestation can be very asymmetrical within the same patient and variable among individuals from the same family. The clinical course is also variable from nonprogressive or slowly progressive over a patient’s lifetime to rapidly progressive with total retinal detachment occurring at a young age.
Three major classifications of FEVR, based on different criteria, have been proposed (Table 1). None of these classifications is without limitations. Gow and Oliver3 suggested a three-stage classification based purely on funduscopic examination. Miyakubo et al4 emphasized the vascular changes in FEVR and included fluorescein angiographic findings in the grading system. Pendergast and Trese5 proposed a system similar to the International Classification for Retinopathy of Prematurity (ICROP) with five stages in the progression of the disease.
The diagnosis of FEVR can be challenging because of its genetic heterogeneity and variable expressivity. Recent identification of several genes involved in FEVR has led to genetic testing that, in our patient, cinched the diagnosis of this condition. The protein products of these genes are all components of the signaling pathway involved in vessel formation of the eye and ear. Mutations in the Norrie disease (NDP)6 gene are associated with X-linked recessive FEVR, mutations in either the Frizzled4 (FZD4)7 or the lipoprotein receptor-related protein 5 (LRP5)8 genes are associated with the autosomal dominant variant, and LRP5 mutations are identified in the autosomal recessive inheritance pattern. Once a diagnosis is made, screening of all family members should be performed.
Despite the universal finding of peripheral avascular zone, not all eyes with FEVR require treatment. Peripheral neovascularization and retinal detachments are the main indication for treatment. Pendergast and Trese suggest the following treatment approach.5
Stage 1: observation only
Stage 2 A and B: laser photocoagulation or cryopexy alone.
Stage 3A: scleral buckle with laser photocoagulation,
Stages 3B, 4B, and 5: scleral buckle, vitrectomy and laser photocoagulation.
Regardless of the stage of disease, all affected patients should be followed closely to monitor for progression.
Table 1
Gow and Oliver
Stage 1: white with or without pressure, cystoid degeneration, vitreous bands, and vitreoretinal traction peripherally
Stage 2: neovascularization in the temporal periphery associated with subretinal exudates and traction resulting in dragging of the disc and major retinal vessels.
Stage 3: total retinal detachment with or without massive subretinal exudates, cataract, iris atrophy, neovascular glaucoma, and band keratopathy.
Miyakubo et al
Type 1: Avascular zone < 2DD from the ora. No neovascularization
Type 2: Avascular zone > 2DD from the ora with well-developed arteriovenous shunt at the peripheral margin.
Type 3: Avascular zone > 2DD with a V-shaped zone along the temporal meridian.
Type 4: Neovascularization at the margin.
Type 5: Presence of solid cicatricial mass at the pars plana.
Pendergast and Trese
Stage 1: Avascular retinal periphery without extraretinal vascularization
Stage 2: Avascular retinal periphery with extraretinal vascularization
A.without exudation
B.with exudation
Stage 3: Retinal detachment-subtotal, not involving fovea
A.primary exudative
B.primary tractional
Stage 4: Retinal detachment-subtotal,
Involving fovea
A.primary exudative
B.primary tractional
Stage 5: Retinal detachment-total
A.open funnel
B.closed funnel
Take Home Points
- Familial exudative vitreoretinopathy is a vitreoretinal dystrophy with variable inheritance, high penetration, and a wide-range of clinical manifestation.
- Treatment for familial exudative vitreoretinopathy is limited to patients with neovascularization and retinal detachments.
- All affected patients should be followed closely to monitor for progression.
- Family members should undergo screening.
Want to Subscribe to Case of the Month?
References
- Criswick VG, Schepens CL. Familial exudative vitreoretinopathy. Am J Ophthalmol. 1969;68:578–594.
- Canny CLB, Oliver GL. Fluorescein angiographic findings in familial exudative vitreoretinopathy. Arch Ophthalmol. 1976;94:1114–1120.
- Gow J, Oliver GL. Familial exudative vitreoretinopathy: an expanded view. Arch Ophthalmol. 1971;86:150–155.
- Miyakubo H, Hashimoto K, Miyakubo S. Retinal vascular pattern in familial exudative vitreoretinopathy. Ophthalmol 1984;91:1524-30.
- Pendergast SD, Trese MT. Familial exudative vitreoretinopathy : Results of surgical management. Ophthalmol 1998;105:1015-23.
- Chen ZY, Battinelli EM, Fielder A, et al. A mutation in the Norrie disease gene (Ndp) associated with X-linked familial exudative vitreoretinopathy. Nat Genet. 1993;5:180–183.
- Toomes C, Bottomley HM, Jackson RM, et al. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am J Hum Genet. 2004;74:721–730.
- Jiao XD, Ventruto V, Trese MT, Shastry BS, Hejtmancik JF. Autosomal recessive familial exudative vitreoretinopathy is associated with mutations in LRP5. Am J Hum Genet. 2004;75:878–884.