West Coast Retina
Case of the Month
November, 2010
Presented by Sandeep Randhawa, MD
A 52-year-old African-American woman presented with decreased vision in the left eye after head trauma.

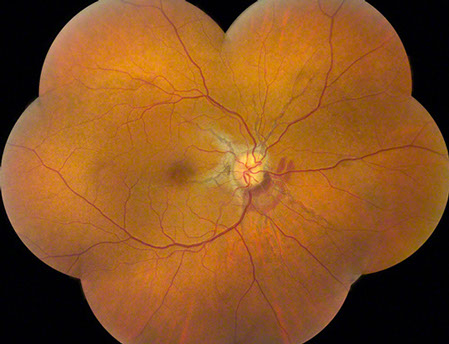
A
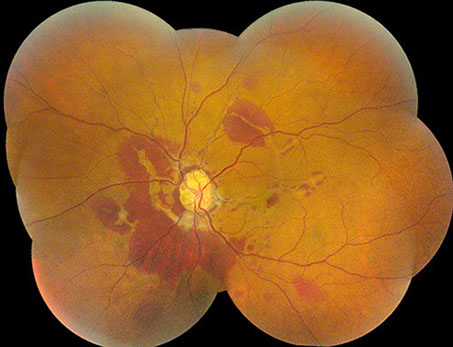
B
Figures 1A and B: Color fundus montages of right and left eye. Note the optic nerve drusen, angioid streaks and subretinal hemorrhage
Case History
A 52-year-old African-American woman presented with decreased vision in the left eye after head trauma. Best-corrected visual acuity measured 20/20 in the right eye and 20/250 in the left eye. Intraocular pressure was 17 mmHg in both eyes. Blood pressure was normal. Fundus examination revealed subretinal hemorrhage and angioid streaks in both eyes (Figs. 1A and B). Prominent optic disk drusen were noted. Fluorescein angiography revealed angioid streaks and choroidal rupture presumed to be related to angioid streaks with extension through the left macula as a result of direct ocular trauma (Figs. 2A and B). B-Scan ultrasonography confirmed the presence of optic nerve head drusen bilaterally (Figs. 3A and B).
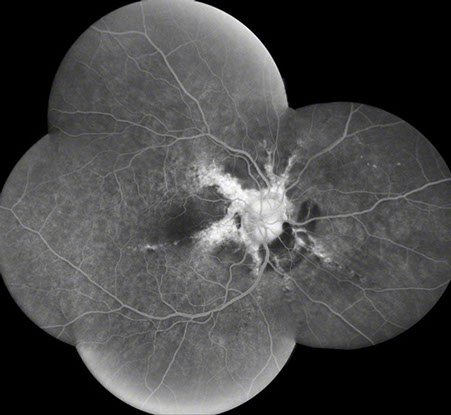
A
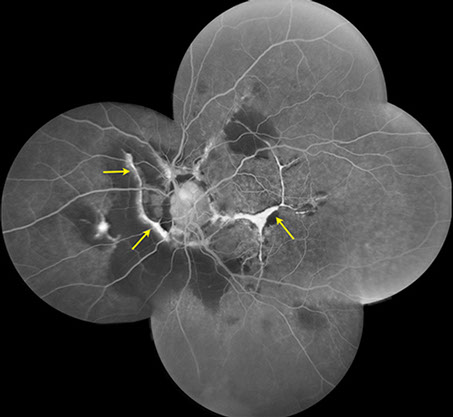
B
Figures 2A and B: Fluorescein angiogram montages of the right and left eye. Note the angioid streaks and subretinal hemorrhage in both eyes, and the choroidal rupture in the left eye (arrows)
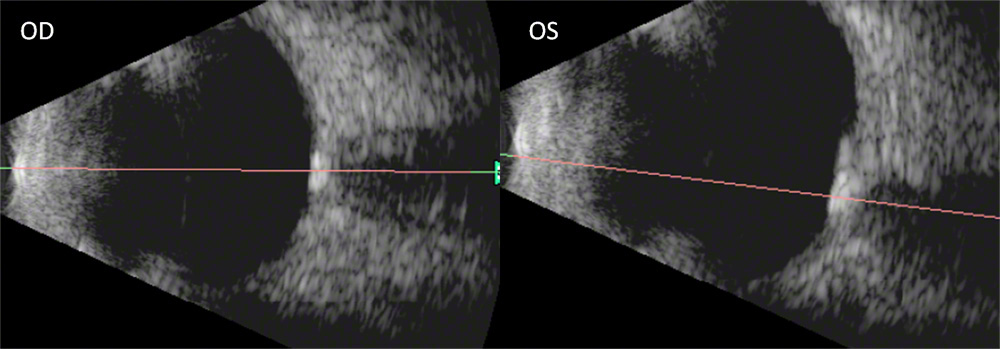
Fig. 3: B-scan ultrasound of the right and left eye showing increased reflectivity on the optic nerve head and increased shadowing consistent with optic nerve drusen
What is your Diagnosis?
Differential Diagnosis
First described by Doyne1 in 1889, angioid streaks are breaks in Bruch’s membrane that originate at the optic disk and radiate out toward the periphery. Most angioid streaks are idiopathic, with 50% of cases associated with a specific systemic disease.2 The most common systemic disease associated with angioid streaks is pseudoxanthoma elasticum, an inherited connective tissue disorder characterized by xanthoma-like papular and reticulated skin lesions.2,3 Other connective tissue disorders associated with angioid streaks include Ehlers-Danlos syndrome and senile elastosis. Hematologic diseases have also been found to be associated with angioid streaks including the sickle cell hemoglobinopathies, thalassemia, acanthocytosis, and hereditary spherocytosis.2 Bony diseases such as Paget’s disease can be associated with angioid streaks as well, in which bony deformities are associated with pain, elevated serum alkaline phosphatase levels, and calcified sclerotic bones.
Clinical Course
Physical examination disclosed inflammatory arthritis of the hands. There were no physical findings suggestive of pseudoxanthoma elasticum or Ehlers-Danlos syndrome. Skin biopsy revealed no evidence of pseudoxanthoma elasticum. Rheumatologic evaluation showed the proximal interphalangeal and distal interphalangeal joints to be affected and also revealed soft-tissue calcific deposits in her hands and wrists that were painful to touch. Radiography of the hands showed erosive osteoarthritis of the proximal interphalangeal and distal interphalangeal joints. Spinal radiography demonstrated intradisk calcification as well as degenerative changes. Radiography of the left hip showed soft-tissue calcification and surrounding edema.
Laboratory evaluation revealed an elevated serum phosphate level of 5.8 mg/dL (normal range, 2.4–4.5 mg/dL) and a slightly elevated erythrocyte sedimentation rate of 52 mm/h. Serum calcium, ionized calcium, and parathyroid hormone levels were within normal limits. Results of tests for antinuclear antibody, anticentromere antibody, and rheumatoid factor were negative. Hemoglobin electrophoresis showed no evidence of sickle-cell disease. Results of radionuclide bone scanning were normal, and bone-specific alkaline phosphatase levels were normal, making a diagnosis of Paget’s disease unlikely.
Endocrinologic evaluation resulted in further examination of the subcutaneous lesions, radiographs, and laboratory data, which led to a diagnosis of familial tumoral calcinosis (FTC). The serum phosphate level was determined again and remained elevated at 5.5 mg/dL. The patient was initially treated with colchicines, but this was of little benefit. She was subsequently treated with prednisone (5 mg daily).
Discussion
Familial tumoral calcinosis (FTC) is a disorder resulting in periarticular calcium deposition, most commonly around the hip, elbow, shoulder, foot, and wrist. The disease is characterized by ectopic soft-tissue calcifications and elevated serum phosphate levels, although FTC has been reported with serum phosphate levels in the normal range.4 Levels of serum calcium and parathyroid hormone are within normal limits. Serum 1,25-dihydroxyvitamin D levels may be inappropriately normal or elevated. Patients generally present within the first two decades of life. Although autosomal dominant pedigrees have been reported,5 most genetic studies favor an autosomal recessive mode of transmission.6 Treatment of FTC is primarily with surgical excision. Elevated phosphate levels in FTC appear to respond to phosphate deprivation (with aluminum hydroxide) in combination with acetazolamide.7
Ocular manifestations of FTC have previously been reported to include palpebral conjunctival calcific nodules, the white limbal girdle of Vogt, calcific band keratopathy, disk drusen, and angioid streaks.11,12 Our patient was not noted to have any external ocular manifestations of FTC but did have angioid streaks, optic nerve head drusen, and the classic rheumatologic and soft-tissue changes of FTC. It is unusual for a patient with FTC to present this late in life, but it appeared that she had been tolerating her arthritis and painful cutaneous deposits for some time before presenting with vision loss. The diagnosis of hyperphosphatemic FTC was made based on the clinical and radiographic presence of soft-tissue calcinosis in an appropriate distribution, elevated serum phosphate levels, and normocalcemia. Although rare, FTC should be considered in the differential diagnosis of angioid streaks, especially when associated with optic nerve head drusen described primarily in families of Jewish Yemenite origin. Ocular manifestations of FTC have previously been reported to include palpebral conjunctival calcific nodules, the white limbal girdle of Vogt, calcific band keratopathy, disk drusen, and angioid streaks.11,12 Our patient was not noted to have any external ocular manifestations of FTC but did have angioid streaks, optic nerve head drusen, and the classic rheumatologic and soft-tissue changes of FTC. It is unusual for a patient with FTC to present this late in life, but it appeared that she had been tolerating her arthritis and painful cutaneous deposits for some time before presenting with vision loss. The diagnosis of hyperphosphatemic FTC was made based on the clinical and radiographic presence of soft-tissue calcinosis in an appropriate distribution, elevated serum phosphate levels, and normocalcemia.
FTC is characterized into subtypes based on levels of serum phosphate. Hyperphosphatemic FTC has been linked to bi-allelic mutations in the UDP-N-acetyl--Dgalactosamine:polypeptide N-acetylgalactosaminyl transferase 3 gene, which encodes a glycosyltransferase that mediates mucin-type O-glycosylation.8 Hyperphosphatemic FTC has also been linked to loss of function mutations in fibroblast growth factor 23, which encodes a protein responsible for increasing urinary phosphate excretion. Gain of function mutations in fibroblast growth factor 23 cause autosomal dominant hypophosphatemic rickets, a disease that is essentially the metabolic mirror image of hyperphosphatemic FTC. It has been suggested that extracellular excretion of fibroblast growth factor 23 may be dependent upon UDP-N-acetyl-µ-D-galactosamine:polypeptide N-acetylgalactosaminyl transferase 3–mediated O-glycosylation.9,10 Normophosphatemic FTC has now been linked to homozygous mutation in the sterile α motif domain–containing protein 9 gene, which is thought to be involved in the regulation of extraosseous calcification.4 The incidence of hyperphosphatemic FTC appears to be higher among patients of African descent, while normophosphatemic FTC has been described primarily in families of Jewish Yemenite origin.
Take Home Points
- Although rare, FTC should be considered in the differential diagnosis of angioid streaks, especially when associated with optic nerve head drusen.
- The findings of angioid streaks merits a medical evaluation as 50% of patients will have an associated systemic disease.
- Patients with angioid streaks are at greater risk of ocular damage with eye trauma and should be advised to wear eye protection.
Want to Subscribe to Case of the Month?
References
- Doyne RW. Choroidal and retinal changes: the result of blows on the eyes. Trans Ophthalmol Soc UK 1889;9:128.
- Mansour AM. Systemic associations of angioid streaks. Int Ophthalmol Clin 1991;31:61–68.
- Clarkson JG, Altman RD. Angioid streaks. Surv Ophthalmol 1982;26:235–246.
- Topaz O, Indelman M, Chefetz I, et al. A deleterious mutation in SAMD9 causes normophosphatemic familial tumoral calcinosis. Am J Hum Genet 2006;79:759–764.
- Lyles KW, Burkes EJ, Ellis GJ, et al. Genetic transmission of tumoral calcinosis: autosomal dominant with variable clinical expressivity. J Clin Endocrinol Metab 1985;60:1093–1096.
- Ichikawa S, Lyles KW, Econs MJ. A novel GALNT3 mutation in a pseudoautosomal dominant form of tumoral calcinosis: evidence that the disorder is autosomal recessive. J Clin Endocrinol Metab 2005;90:2420–2423.
- Yamaguchi T, Sugimoto T, Imai Y, et al. Successful treatment of hyperphosphatemic tumoral calcinosis with longterm acetazolamide. Bone 1995;16:247S–250S.
- Topaz O, Shurman DL, Bergman R, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet 2004;36:579–581.
- Benet-Pages A, Orlik P, Strom TM, et al. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet 2005;14:385–390.
- Larsson T, Yu X, Davis SI, et al. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab 2005;90:2424–2427.
- Bruns DE, Lieb W, Conway BP, et al. Band keratopathy and calcific lid lesions in tumoral calcinosis. Case reports. Arch Ophthalmol 1988;106:725–726.
- Ghanchi F, Ramsay A, Coupland S, Barr D, Lee WR. Ocular tumoral calcinosis. A clinicopathologic study. Arch Ophthalmol 1996;114:341–345.