West Coast Retina
Case of the Month
October, 2011
A 29-year-old male with sudden loss of vision in both eyes.
Presented by Sandeep Randhawa, MD

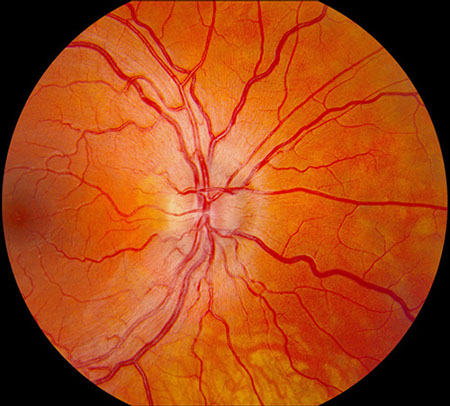
A
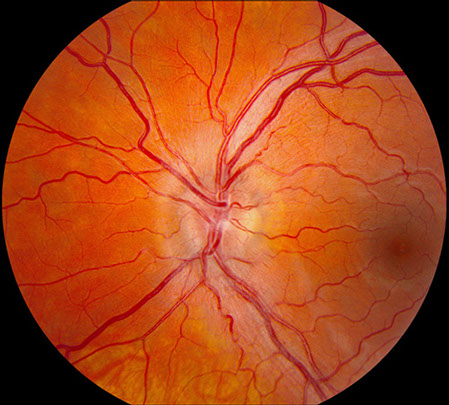
B
Figures 1A and B: Fundus photograph of the right and left eye. Note the swelling of the optic disc in each eye. Mild telangiectatic changes are seen at the disc margins and mild retinal vascular tortuosity is present. The macula in both eyes is normal.
Case History
A 29-year-old male presented with a two-week history of acute visual loss in both eyes. This was associated with headaches for which he had been taking aspirin. His past medical history and family history were unremarkable.
On examination, best-corrected visual acuity was 20/200 both eyes. There was no relative afferent pupillary defect. The intraocular pressure and anterior segment examination was normal in both eyes. On Ishihara color vision testing, he could only identify 6 of 14 plates correctly in both eyes with responses indicating a red-green dyschromatopsia. Fundus examination revealed bilateral optic disc swelling, fine telangiectatic vessels at the disc margins, and mild vascular tortuosity (Figure 1). The macula and peripheral retina were normal in both eyes.
What is your Diagnosis?
Differential Diagnosis
The differential diagnosis of a bilateral optic neuropathy includes papilledema, bilateral optic neuritis, anterior compressive optic neuropathy, infectious optic neuropathy, infiltrative optic neuropathy, toxic/nutritional optic neuropathy, ischemic optic neuropathy, and hereditary optic neuropathy. In papilledema, the visual acuity is initially normal, and the visual field may be normal or may demonstrate an enlargement of the blind spot. Optic neuritis is typically associated with ocular discomfort aggravated by eye movement, and recovery of vision from optic neuritis usually begins after 1 or 2 weeks. Orbital lesions compressing the optic nerve (e.g. Graves myopathy or orbital tumors), infiltrative disease of the optic nerve (e.g. lymphoma, metastatic carcinoma, or sarcoidosis) or infections of the optic nerve (cryptococcus or cytomegalovirus) may cause disc edema and visual loss. The duration of vision loss is typically less rapid in these disorders and clinical findings, such as proptosis, a history of immunosuppression, or the presence of a known primary cancer are often present. Anterior ischemic optic neuropathy is uncommon in patients less than 50 years old and is associated with altitudinal visual field defects. Toxic or nutritional optic neuropathies are notable for a normal optic disc or mild temporal pallor. Hereditary optic neuropathies include Leber’s hereditary optic neuropathy (LHON) affecting males between 10 and 30 years of age, and Kjer dominant optic atrophy, affecting both males and females between 4 to 8 years of age.
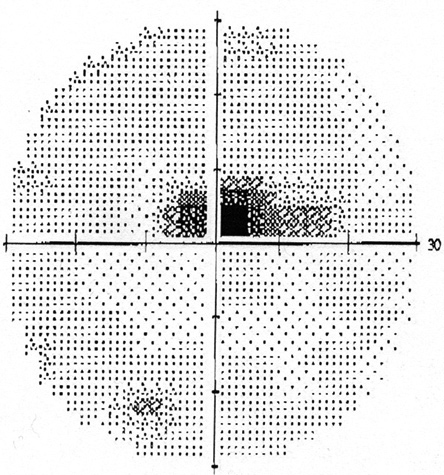
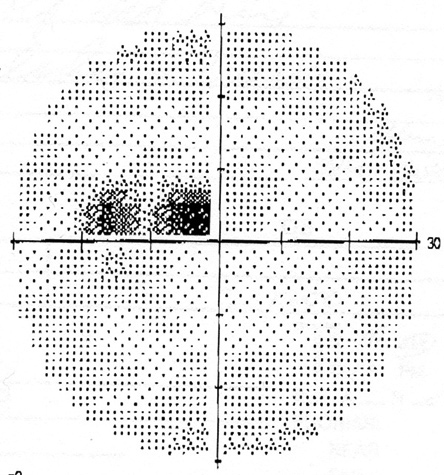
Figure 2: Humphrey visual field tests of both eyes. A cecocentral scotoma is present in each eye.
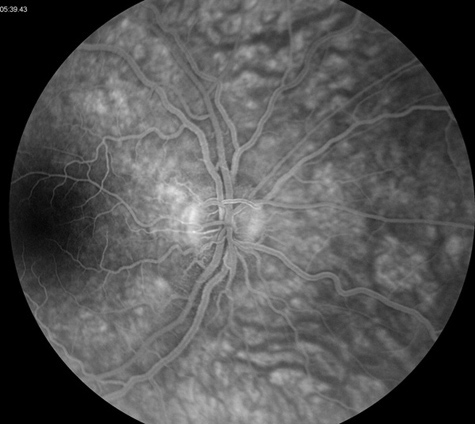
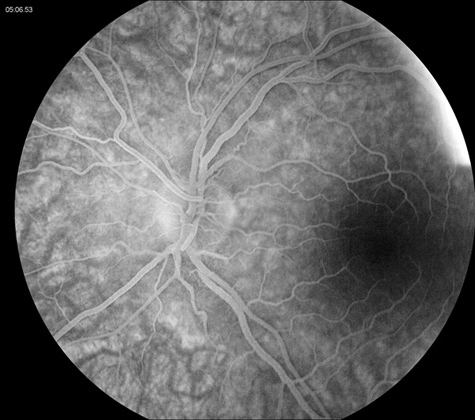
Figure 3: Fluorescein angiogram of the right and left eye. Note the absence of disc leakage. The macula is normal in each eye.
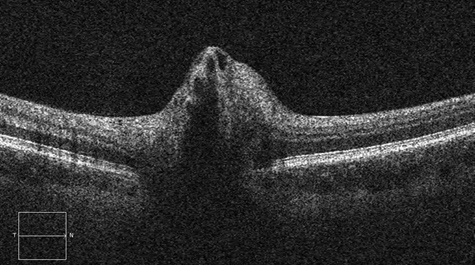
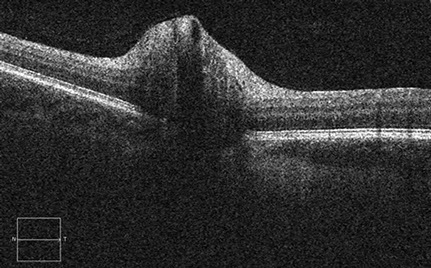
Figure 4: Spectral domain optical coherence tomography of the right and left nerves. Peripapillary nerve fiber layer swelling is present in each eye
Clinical Course
Humphrey visual field test revealed a cecocentral scotoma in both eyes (Figure 2). Fluorescein angiography revealed no leakage from the optic disc (Figure 3). Optical coherence tomography of the macula was normal, but confirmed peripapillary nerve fiber layer swelling in both eyes (Figure 4). A magnetic resonance imaging scan of the brain and orbit was normal.
Based on the symptoms of sudden painless visual loss, optic disc edema with circumpapillary telangiectatic microangiopathy, mild vascular tortuosity, and absence of leakage from the disc, a diagnosis of Leber’s hereditary optic neuropathy was suspected. Mitochondrial testing confirmed a mitochondrial DNA mutation at nucleotide position 11778 (Guanine to Adenosine) in the ND4 gene responsible for LHON.
Discussion
Leber’s hereditary optic neuropathy typically affects males between 10 to 30 years of age but can occur much later in life. Women may account for 10%–20% of reported cases. The syndrome often presents with severe, acute, painless, visual loss typically worse than 20/200 with an associated central or cecocentral visual field scotoma.1 The classic fundus appearance includes:
•Hyperemia and elevation of the optic disc with thickening of the peripapillary retina
•Peripapillary telangiectasia
•Absence of disc leakage on fluorescein angiography
These findings may be present before visual loss begins.2 Though typically painless, LHON may be associated with headache as well as transient worsening of vision with warmth or exercise (Uhthoff's phenomenon), photopsia, limb paresthesia, and dizziness.2 The classic triad of circumpapillary telangiectatic microangiopathy, swelling of the nerve fiber layer around the disc, and absence of leakage from the disc on fluorescein angiography may be observed in 50% to 60% of affected patients.2-3 The fundus may be entirely normal in up to 40% of patients.2
Simultaneous, bilateral vision loss is reported in approximately 50% of cases. In eyes with initial unilateral involvement, the average time to fellow eye involvement is 3 months but has been reported as late as 16 years. With time, hyperemia of the disc and peripapillary nerve fiber layer swelling resolve, leaving temporal pallor and papillomacular nerve fiber layer atrophy. Although visual loss is typically permanent, partial recovery of vision, even years after initial loss, has been reported in 10% to 20% of cases.4-5
LHON is the associated with a mitochondrial DNA mutation, most frequently at the 11778 position, and less commonly at the 3460 or 14484 positions. The corresponding single base-pair nucleotide substitution results in impaired mitochondrial adenosine triphosphate production, which affects highly energy-dependent tissues such as the optic nerve. Genotypic confirmation aids in genetic counseling and may provide information about prognosis. Patients with the 14484 mutation have a higher chance (65%) of late spontaneous improvement in central visual function, whereas those with the 11778 mutation have a lower chance (4%).4-5
The mode of transmission of mitochondrial DNA disorders is maternal and not from nuclear DNA. Thus, only women transmit the disease. Not all men with affected mitochondria experience visual loss, and affected women develop visual symptoms only infrequently. The reason for selective male susceptibility is unclear, as is the precipitating event. LHON exhibits heteroplasmy, in which a variable percentage of DNA is affected, and only when this percentage becomes high does the disease become clinically evident. Family history may be difficult to elicit, and in a significant number of cases there is a negative family history suggesting a de novo mutation.4-5
Occasionally, patients demonstrate cardiac conduction abnormalities or other mild neurologic deficits that warrant further evaluation. No treatment has been shown to be effective. Corticosteroids are not beneficial. Coenzyme Q10, succinate, and other agents that may increase mitochondrial energy production have been proposed, but no definite benefit has been shown. Avoidance of agents such as tobacco or excess ethanol, which might stress such energy production, is recommended, though definitive studies are lacking.
Take Home Points
- LHON may present in the absence of a known family history.
- The classic triad of LHON is sudden profound painless visual loss, disc edema with circumpapillary telangiectatic microangiopathy, and absence of disc leakage on fluorescein angiography.
- Genetic counseling is important. Males with a LHON mutation will not pass the mutation or the disease to their children. Females with known LHON mutations will pass the mutation to males and females, although not all persons with the mutation will become symptomatic.
Want to Subscribe to Case of the Month?
References
- Newman NJ. Leber’s hereditary optic neuropathy: new genetic considerations. Arch Neurol.1993;50:540–548.
- Newman NJ. Leber’s hereditary optic neuropathy. Ophthalmol Clin North Am.1991;4:431–447.
- Nikoskelainen E, Hoyt WF, Nummelin K, et al. Fundus findings in Leber’s hereditary optic neuroretinopathy. III. Fluorescein angiographic studies. Arch Ophthalmol. 1984;102:981–989.
- Nikoskelainen EK, Huoponen K, Juvonen V, et al. Ophthalmologic findings in Leber hereditary optic neuropathy, with special reference to mtDNA mutations. Ophthalmology.1996;103:504–514.
- Riordan-Eva P, Sanders MD, Govan GG, et al. The clinical features of Leber’s hereditary optic neuropathy defined by the presence of a pathogenetic mitochondrial DNA mutation. Brain.1995;118:319–337.