West Coast Retina
Case of the Month
September, 2012
A 26-year-old man with long history of reduced vision in his right eye.
Presented by Paul Stewart, MD

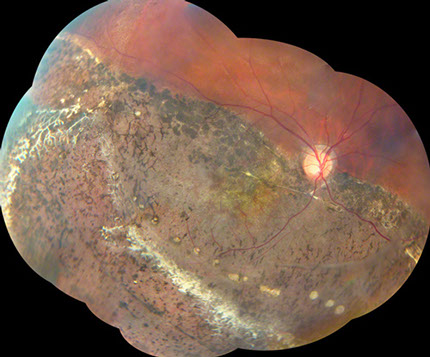
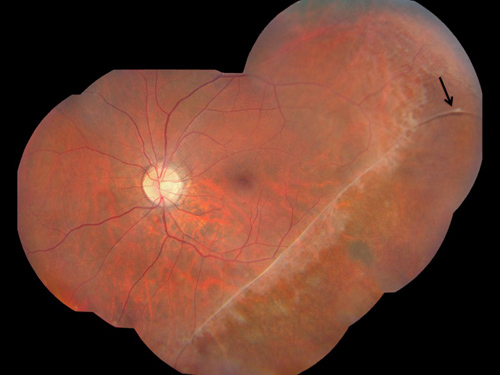
Figure 1: Color montage of the right eye shows widespread pigment disruption and atrophy from 4 to 10:00 with dendritic pruned vessels in the inferotemporal region.
Figure 2: Color montage of the left eye shows an elevated band with vessel at 3:00 in the periphery (arrow). There is a band of fibrosis inferotemporally with the granularity of the RPE more visible anteriorly
Case History
A 26-year-old man was referred for evaluation of long-standing decreased vision in the right eye. He could not remember a time when the vision in that eye was normal. He had no change in vision and felt the vision in his left eye was good. He had a large angle exotropia that was present his whole life. He had no significant past medical or surgical history. There was no family history of any eye disease.
On examination vision in the right eye was 3/200 and in the left was 20/20. His intraocular pressures were 14 and 12 in the right and left eyes respectively. Anterior segment examination of both eyes was normal. Fundus examination of the right eye showed pigment epithelial disruption and clumping extending from four to ten o’clock and involving the macula (Figure 1). There were areas of dendritic, sheathed vessels in the periphery. Fundus examination of the left eye showed a band of fibrosis inferotemporally with an elevated band at three o’clock (Figure 2). The inferotemporal region had much more visible underlying RPE. The fluorescein angiogram of the right eye showed widespread window defects corresponding to the areas of atrophy seen on the color photographs (Figure 3). The left eye angiogram showed the fovea to be above the horizontal midline (Figure 4).
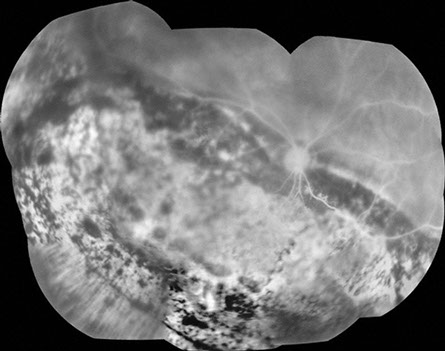
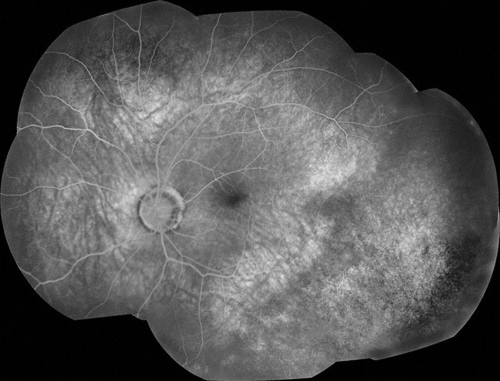
Figure 3: Fluorescein angiography of the right eye shows widespread window defects corresponding to areas of retinal pigment epithelial atrophy. Significant areas of hypofluorescence are present as well due to the retinal pigment epithelial hyperplasia.
Figure 4: Fluorescein angiography of the left eye shows the fovea to be displaced above the horizontal midline. The inferotemporal region appears poorly perfused.
What is your Diagnosis?
Differential Diagnosis
The differential diagnosis includes X-linked retinoschisis (XLRS), Goldmann-Favre syndrome, congenital stationary night blindness, degenerative retinoschisis, Stickler syndrome, retinitis pigmentosa and familial exudative vitreoretinopathy.
Clinical Course
Genetic testing was performed confirming the diagnosis of X-linked retinoschisis. Given the high clinical suspicion, the patient’s blood was sequenced through the entire coding sequence of the RS1 gene, showing a possible high-penetrance disease-causing sequence variation with an amino acid change of Arg102Gln.
The patient’s visual acuity and ocular examination have remained the same over the past eleven years of follow-up. SD-OCT of the right eye showed significant outer retinal atrophy and disorganization with inner retinal cystic changes, but without a schisis. There is diffuse retinal pigment epithelial clumping and atrophy (Figure 5). SD-OCT of the left eye showed normal foveal contour and normal retinal structure except for a loss of distinct lamination between the outer nuclear layer and outer plexiform layers centrally (Figure 6). The posterior hyaloid was incompletely separated. There was no schisis present. Genetic testing has showed a hemizygous nucleotide substitution mutation in the RS1 gene that is likely to be disease causing for X-linked retinoschisis.
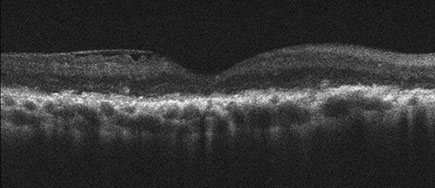
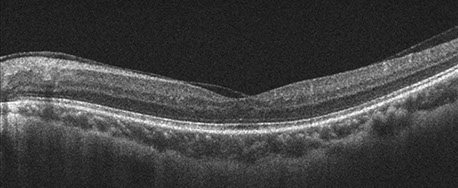
Figure 4: SD-OCT of the right macula shows significant atrophy of the retina with loss of the outer retinal layer architecture, but no retinal schisis. There is pigment clumping and irregular retinal pigment epithelium.
Figure 6: SD-OCT of the left macula shows no retinal schisis in any layer.
Discussion
X-linked juvenile retinoschisis (XLRS) is a retinal dystrophy found in males with variable severity. The first clinical reports were in the 19th century and was first documented as X-linked in 1913.1 It is caused by mutations in the RS1 gene, first identified in 1997, that lead to schisis of the retina both in the fovea and peripherally through abnormalities in the retinoschisin protein. There is a characteristic spoke-wheel pattern formed by the foveal splitting. The foveal schisis can occur in the inner nuclear layer (INL), the outer plexiform layer (OPL), the outer nuclear layer (ONL) and rarely the retinal nerve fiber layer (RNFL).2, 3 Adaptive optics scanning laser ophthalmoscopy has shown that the cone spacing is increased within foveal schisis cavities.4 Foveal schisis is reported to occur in almost all cases of XLRS, but may not be seen in individuals over the age of 30 and rarely is not seen in younger individuals.5, 6 In our patient foveal schisis is not present. Either he is old enough that the foveal schisis is no longer present or he is on the rare end of the spectrum of disease of XLRS where he does not have the foveal findings. Certainly in retinoblastoma, a better studied heritable eye condition, there is a wide range of phenotypes from mild to severe, likely related to the massive variety (over 900 reported) of disease-causing mutations.
Peripheral schisis is seen in 50-70 percent of patients, most commonly inferotemporally. There can be giant inner retinal layer holes, as is seen in the left eye of our patient. Occasionally retinal vessels will be all that remain in the elevated schisis inner layer giving the appearance of vitreous veils. If these vessels break there can be vitreous hemorrhage. As the vessels of the inner retina dive towards the outer retina, they can hemorrhage as the schisis pulls them apart. These vessels can also close and become fibrotic giving a characteristic appearance seen temporally in our patient’s right eye. This configuration of dendritic pruned vessels can also be seen in Goldmann-Favre disease and we have seen very similar vessels in Coat’s disease as well.
Retinal detachment is another potential complication when a patient acquires both inner and outer retinal holes. Surgical repair can be attempted with beneficial outcomes.7 When performing a vitrectomy, particularly in a young patient, it may be necessary to remove the schisis inner wall during the vitrectomy. Our patient’s right eye has the characteristic appearance of a spontaneously resolved retinal detachment. Given his long-standing history of poor vision in this eye, he most likely had this detachment very early in his life.
Take Home Points
- X-linked juvenile retinoschisis classically has a characteristic foveal spoke-like schisis and frequently has peripheral schisis, although neither is a universal finding.
- XLRS also has a characteristic closed-vessel appearance of fibrotic retinal vessels.
- •Retinal detachment and vitreous hemorrhage are complications that may require surgical intervention.
Want to Subscribe to Case of the Month?
References
- Pagenstecher H. Uebereine unterdembildeder Natzhauterblosung verlaufende, erbiche ErkankungderRetina. Graefes Arch Clin Exp Ophthalmol 1913; 86: 457-462.
- Gregori NZ, Berrocal AM, Gregori G, et al. Macular spectral-domain optical coherence tomography in patients with X-linked retinoschisis. Br J Ophthalmol 2009; 93: 373-378.
- Yu J, Ni Y, Keane PA, et al. Foveomacular schisis in juvenile X-linked retinoschisis: an optical coherence tomography study. Am J Ophtho 2010; 149: 973-978.
- Duncan JL, Ratnam K, Roorda A, et al. Abnormal cone structure in foveal schisis cavities in X-linked retinoschisis from mutations in exon 6 of the RS1 gene. IOVS 2011; 52: 9614-9623.
- Eadie JA, Luo CK, Trese MT. Novel clinical manifestation of congenital X-linked retinoschisis. Arch Ophthalmol 2012; 130: 255-257.
- George ND, Yates JR, Moore AT. Clinical features in affected males with X-linked retinoschisis. Arch Ophthalmol 1996; 114: 274-280.
- Rosenfeld PJ, Flynn HW Jr, McDonald HR, et al. Outcomes of vitreoretinal surgery in patients with X-linked retinoschisis. Ophthalmic Surg Las 1998; 29: 190-197.