April, 2023
Presented by Malini Veerappan Pasricha, MD


Presented by Malini Veerappan Pasricha, MD
A 72-year-old Hispanic woman presents with two years of progressive nyctalopia in both eyes.
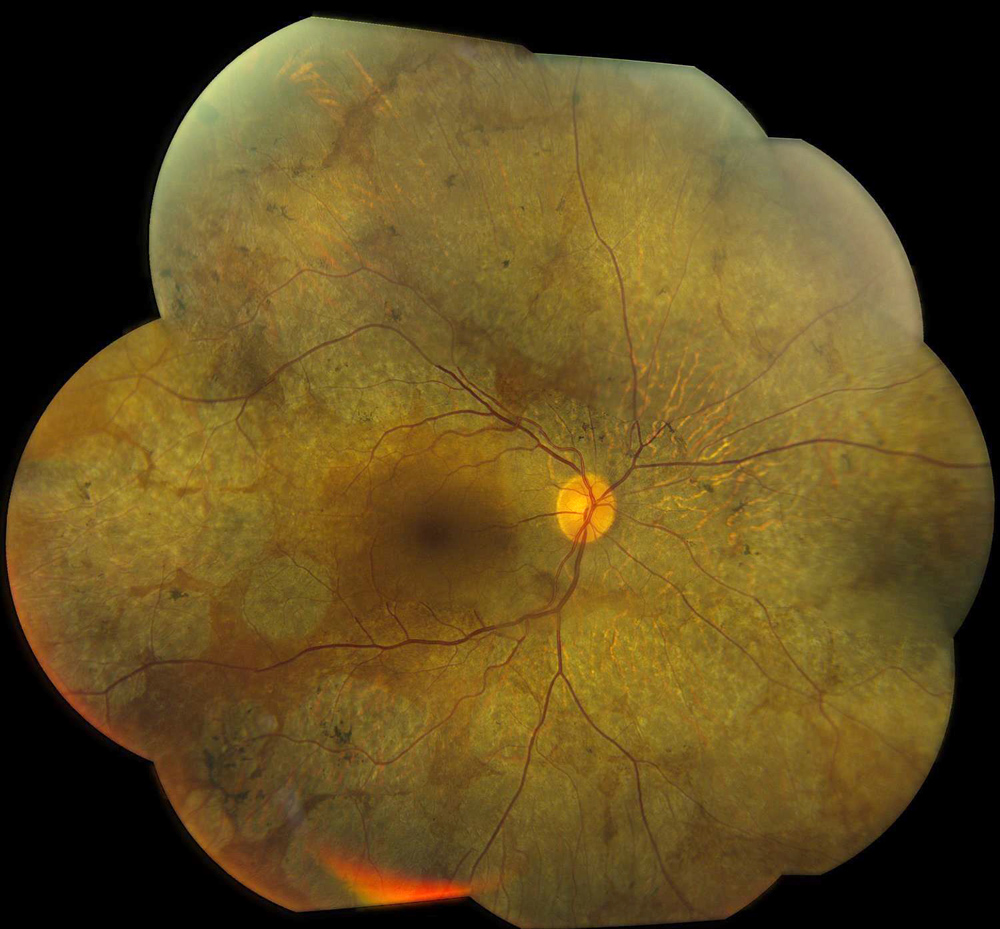
Figure 1A: Color photo montage of the right eye. Note the extensive areas of chorioretinal atrophy extending from the macula to the periphery.
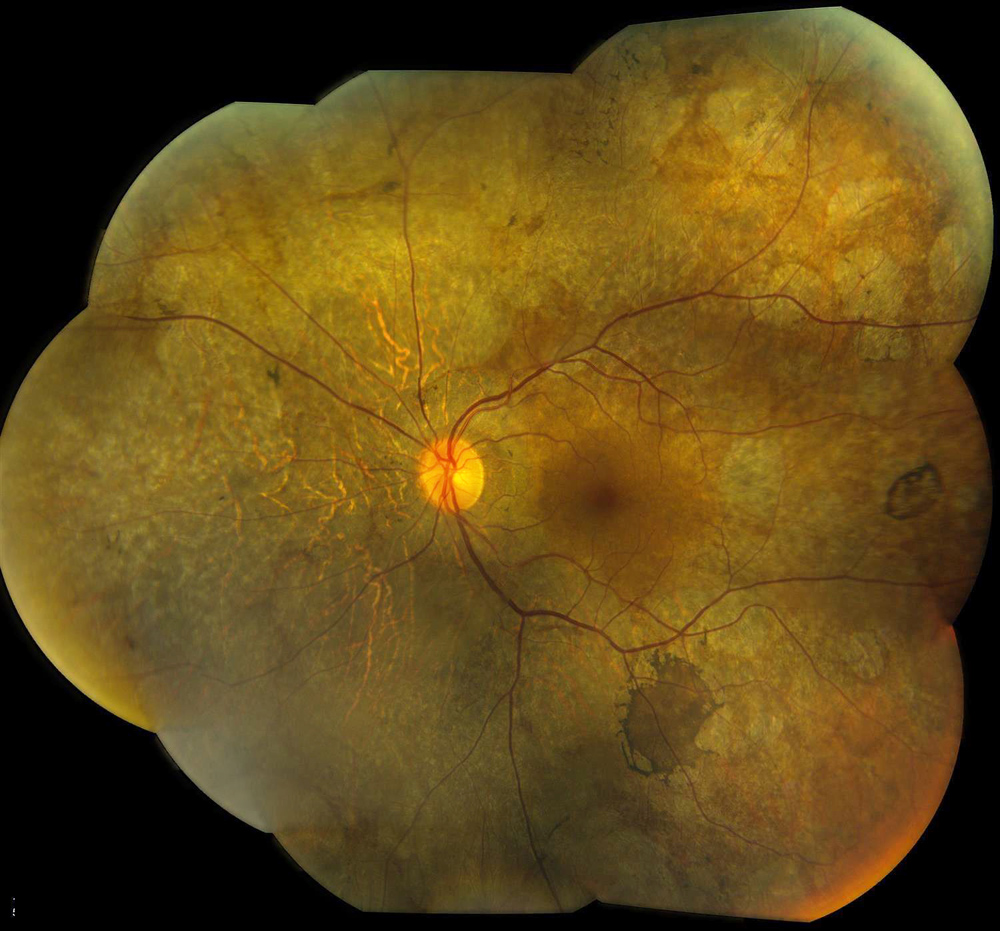
Figure 1B:Color photo montage of the left eye. Note the extensive areas of chorioretinal atrophy extending from the macula to the periphery as seen in the right eye.
She presented with generalized visual decline over the last few months in addition to 2 years of nyctalopia. She denied any flashes, floaters, scotoma, pain, or headaches. Her past ocular history was significant for pseudophakia. Medical history was significant for type 2 diabetes (without ocular complications) and asthma. Surgical history was significant for multiple orthopedic surgeries for lower extremity fractures due to recent falls. Her only medication was insulin. Family history was negative for ocular disorders in her parents and siblings, but she reported that her son had similar visual complaints. No known allergies. She denies use of tobacco or illicit substances. Review of systems was negative.
The patient’s best corrected Snellen visual acuity measured 20/32 OD and 20/50 OS. Intraocular pressures were normal. Anterior segment exam was age-appropriate. The funduscopic exam of both eyes was significant for generalized arteriolar narrowing, pigment clumping in the periphery, and patchy RPE atrophy of the macula and periphery (Figures 1A and 1B). The fluorescein angiogram of both eyes showed transmission defects consistent with severe atrophy of the RPE and choriocapillaris, as well as patchy choroidal filling (Figures 2A and 2B). OCT macula of both eyes showed mild cystoid macular edema and perifoveal outer retinal layer loss (Figures 3A and 3B). Goldmann visual field testing showed dense mid-peripheral scotomas in the right eye and more advanced mid-peripheral loss in the left eye (Figures 4A and 4B). ERG showed loss or delay of all signals, indicative of near total dysfunction of rods and cones (Figure 5).
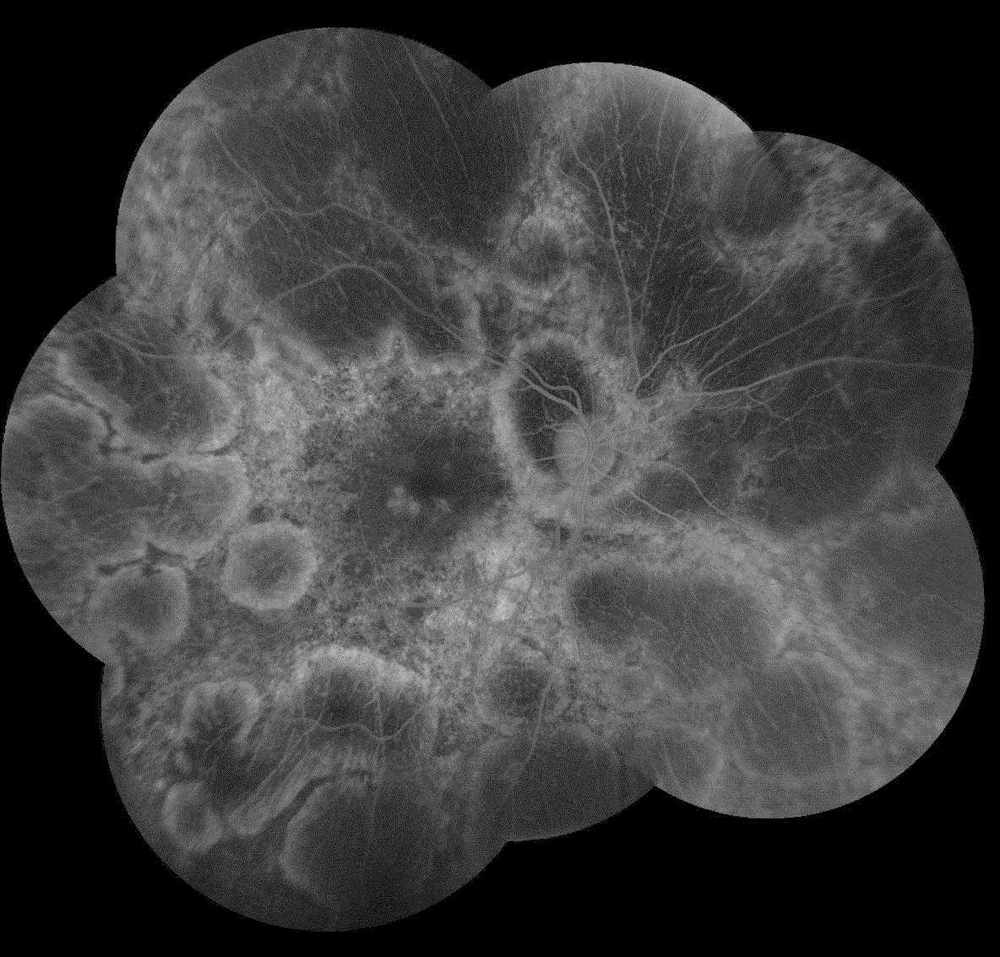
Figure 2A:Fluorescein angiogram montage of the right eye. Note the extensive areas of hypofluorescence due to loss of choriocapillaris and other areas of transmission defects.
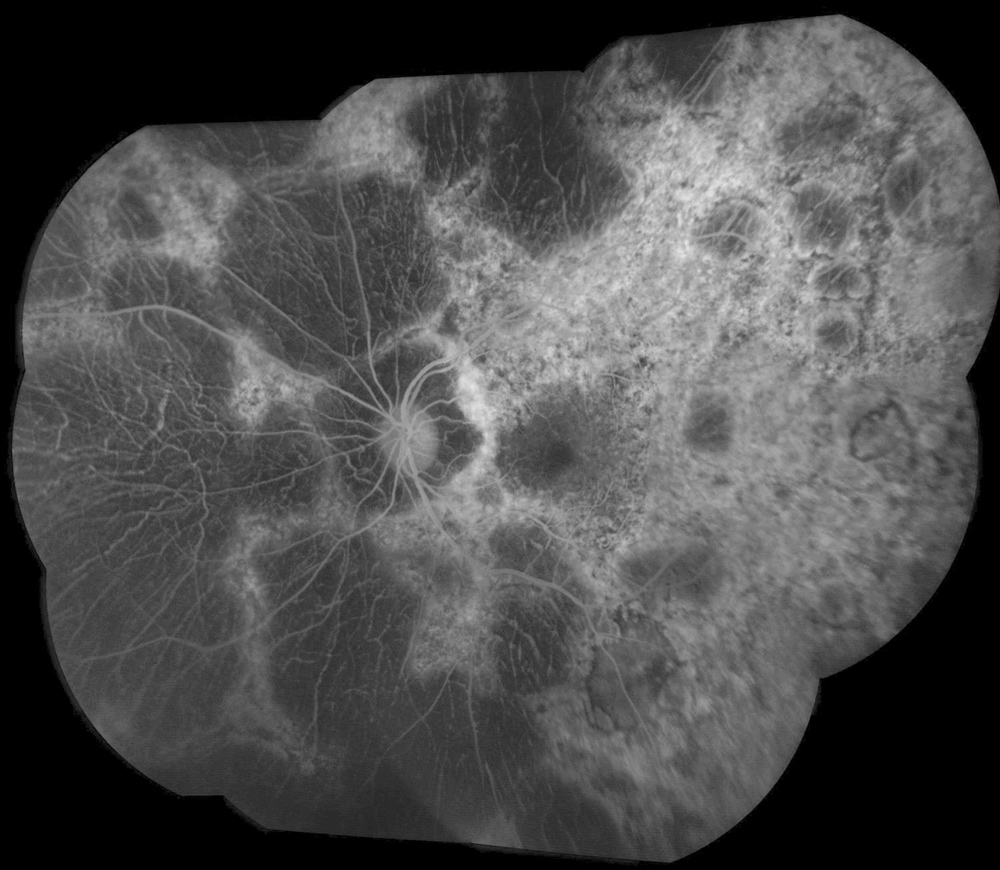
Figure 2B:Fluorescein angiogram montage of the left eye. Similar findings are noted in the left eye as seen in the right. There are extensive areas of hypofluorescence due to loss of choriocapillaris and other areas of transmission defects
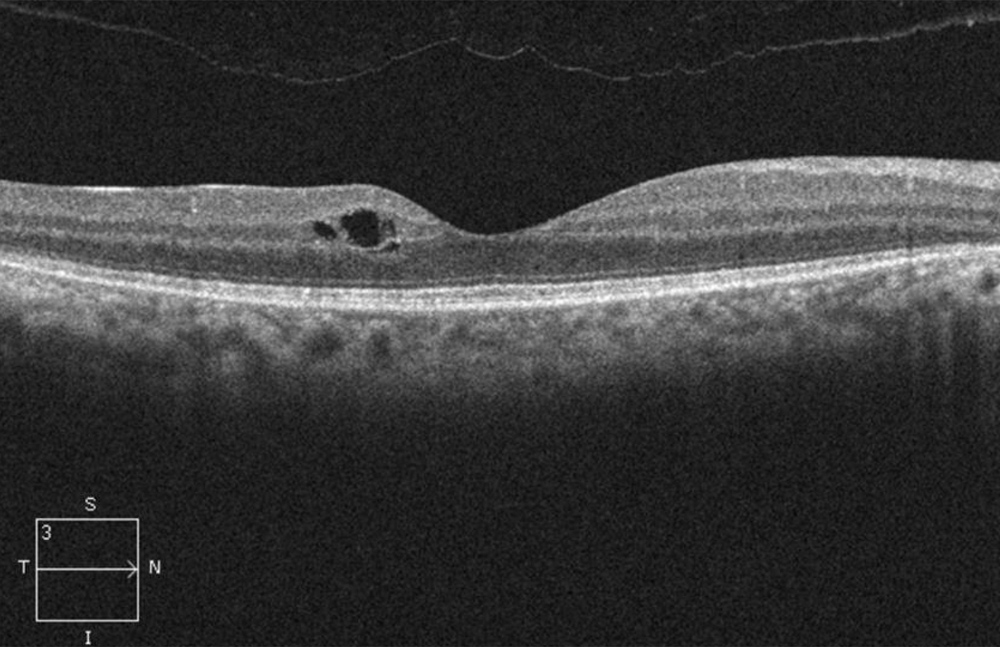
Figure 3A:OCT-SD scan of the right macula. Note mild mcular edema but relative preservation of the retinal layers centrally but outer retinal loss pericentral.
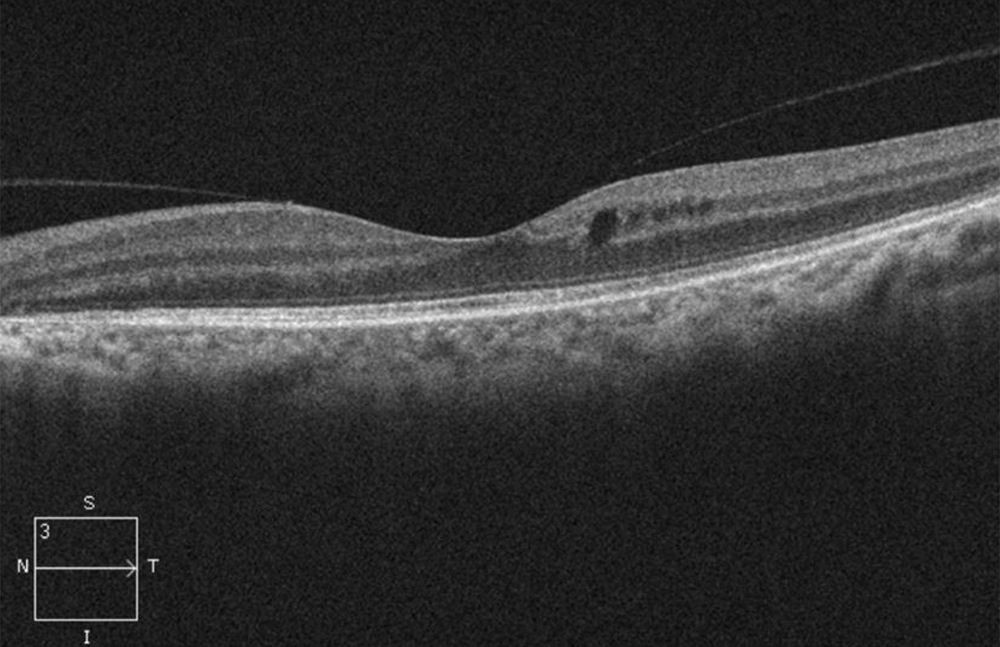
Figure 3B:OCT-SD scan of the left macula. Note mild mcular edema but relative preservation of the retinal layers centrally but outer retinal loss pericentral.
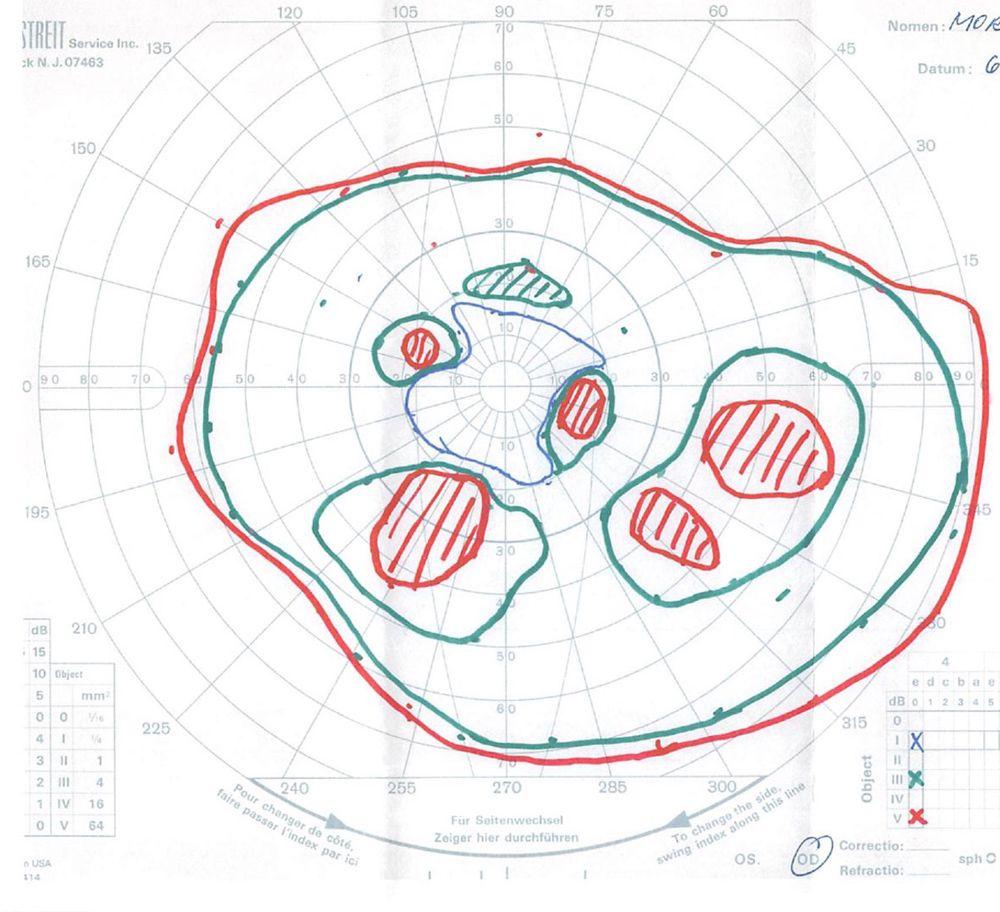
Figure 4A:Goldmann VF of the right eye. Note the dense mid-peripheral scotomas.
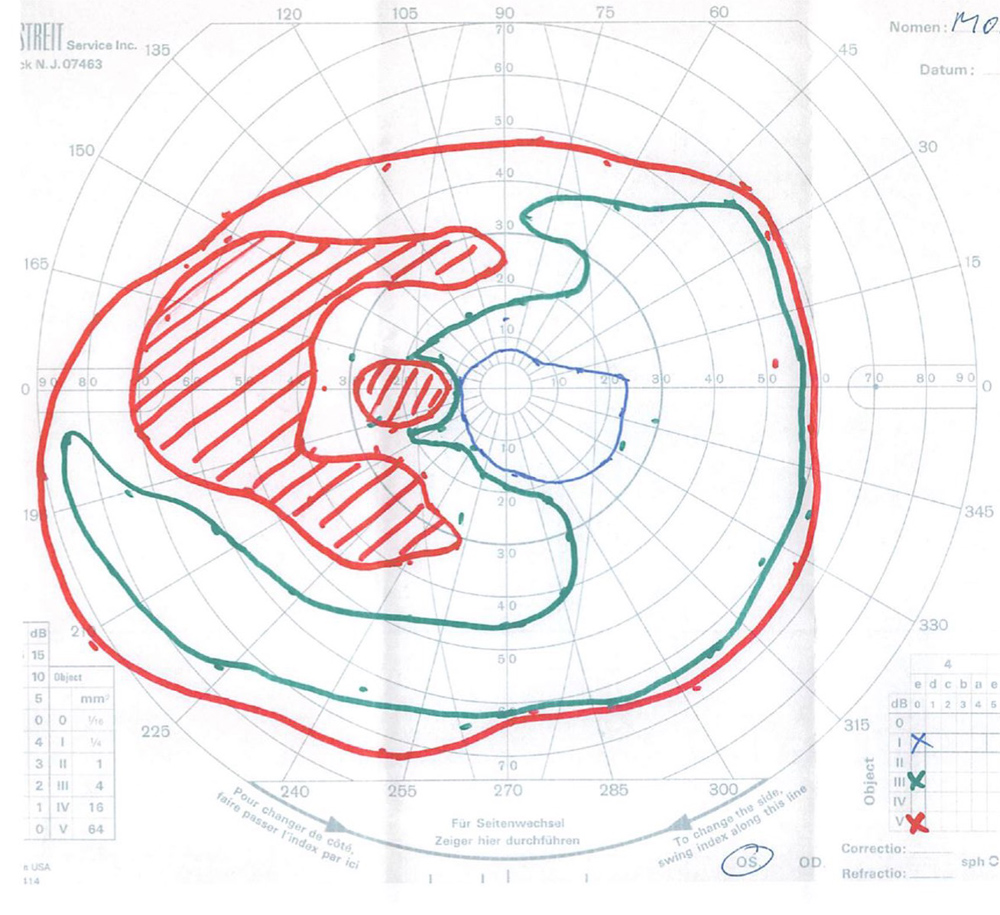
Figure 4B:Goldmann VF of the left eye. Note the even more dense mid-peripheral scotomas in this eye as compared with the right..
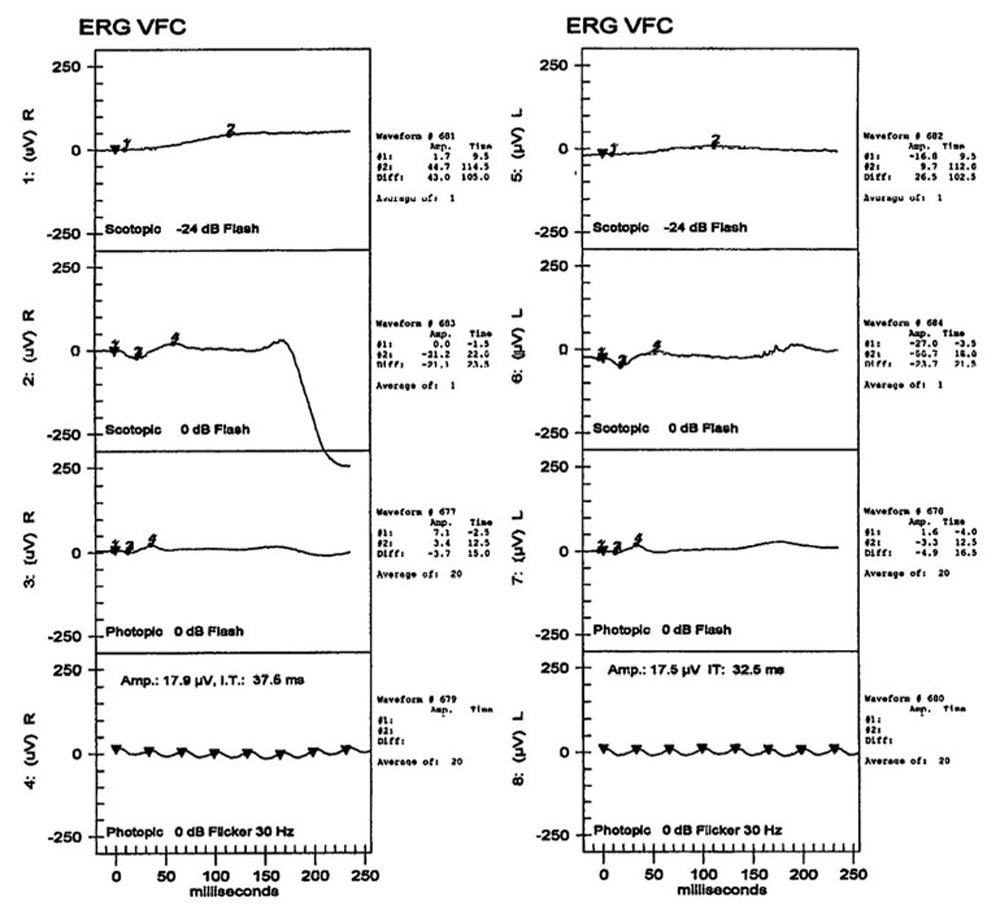
Figure 5:ERG testing of both eyes shows severe loss of rod and cone function.
Differential Diagnosis
Inherited
Acquired
Infectious
Additional History and Patient Course
The patient underwent genetic testing and was found to be a carrier of the CHM mutations for choroideremia (Figures 6A and 6B). No other genetic defects were found. The patient’s vision has declined slowly over the last decade. Her cystoid macular edema has been variably responsive to topical carbonic anhydrase inhibitors and steroids, as well as periocular steroids. We are hoping to evaluate her son given his similar symptoms, to determine if he has any evidence of disease.

Figure 6A:Genetic testing showed her to be heterozygous for a deletion in the CHM gene.
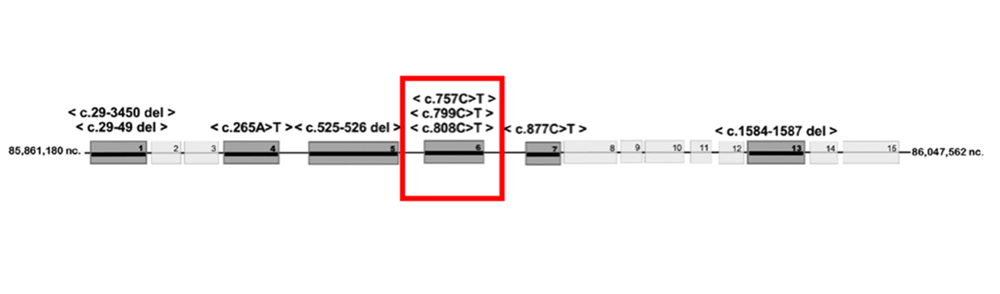
Figure 6B:Genetic testing results.
Discussion
Choroideremia is an X-linked chorioretinal dystrophy characterized by progressive, diffuse degeneration of the RPE, choriocapillaris, and photoreceptors.1 It is estimated to affect between 1 in 50,000 to 1 in 100,000 individuals, with highest prevalence reported in parts of Europe.1,2 Affected patient present with nyctalopia in the first decade of life, progressive loss of peripheral vision through early adulthood, followed by rapid deterioration of color and central vision in the fifth to seventh decades of life.3 There can be striking variations in phenotypic manifestations within the same family.4
On fundus examination, widespread pigment clumping at the level of the RPE - distinct from the characteristic perivascular bone spicules seen in retinitis pigmentosa – is the earliest manifestation of disease. This is followed by well-defined regions of RPE atrophy, most commonly in the post-equatorial region outside of the vascular arcades. These areas of atrophy advance centripetally and eventually involve the foveal center. Scleral and larger choroidal blood vessels become visible with the atrophy, but are anatomically preserved along with the retinal vessels. Unlike retinitis pigmentosa, there is no waxy pallor or atrophy of the optic disc. Other ocular findings in a minority of choroideremia patients include posterior subcapsular cataracts, cystoid macular edema, and choroidal neovascularization (rare).1,5,6
Choroideremia is caused by mutations in the CHM gene, and over 106 pathogenic variations have been identified.6,7 The CHM gene encodes for the Rab-escort-protein-1 (REP-1), which plays a role in the maintenance of RPE cells.8 The diagnosis of choroideremia can be ascertained by family history and fundus findings. Genetic testing or immunoblot analysis with anti-REP-1 antibody is confirmatory.9 Though there is currently no effective treatment, choroideremia is a promising candidate for gene therapy since it is caused by a mutation of one gene. Investigational therapies have focused on replacing the defective gene via viral vectors. Adeno-associated virus subtype 2 (AAV2) has been of special interest as it has a particular affinity for primate photoreceptors and RPE.10
The CHM gene is located on chromosome X and the disease is inherited in an X-linked recessive manner.1 Thus, men are predominantly affected; however, women can be asymptomatic or symptomatic carriers of the disease. Carrier phenotypes can range from mild RPE changes to patchy RPE atrophy.11,12 Lyonization is responsible for this phenotypic variability, in which one of the two copies of the X chromosome in females is silenced during early embryogenesis.5 Female choroideremia carriers have occasionally shown advanced pathology that progresses with age.13,14 Additionally, female carriers with certain X-autosome translocations can present with mild to severe clinical features of choroideremia associated with systemic issues such as ovarian dygenesis or ectodermal dysplasia.6