August, 2020
Presented by Braden Burckhard, MD


Presented by Braden Burckhard, MD
A 45-year-old Caucasian female patient was referred for evaluation of progressively worsening night vision and blurred central vision when transitioning from light to dark environments.
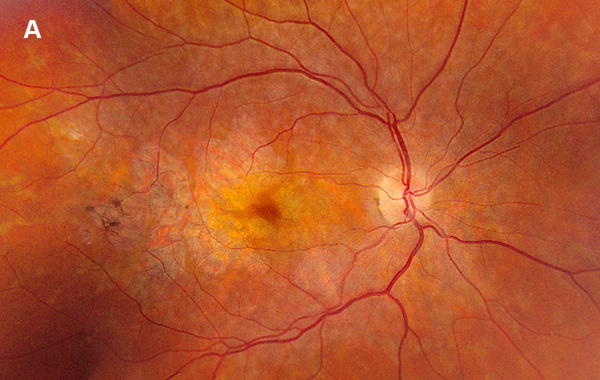
Figure 1A: Color photo of the right eye. Note areas of atrophy temporal to the macula as well as areas of pigment clumping, and RPE thinning in the posterior pole.
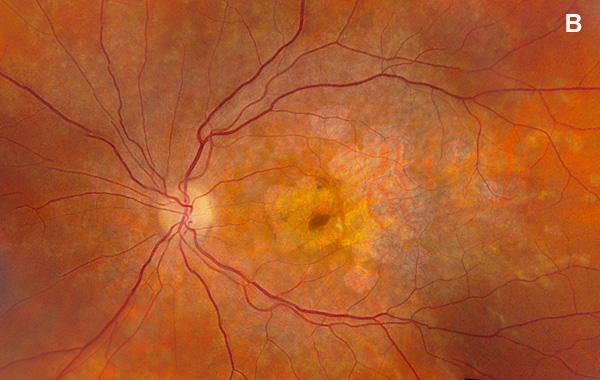
Figure 1B: Color photo of the left eye. Note areas of atrophy temporal to the macula as well as areas of pigment clumping, and RPE thinning in the posterior pole.
A 45-year-old Caucasian female gave a one-year history of increasing vision problems. She noted difficulty transitioning from light to dark, glare and light sensitivity. Also, she reported ‘white blotches’ in her vision affecting her ability to recognize faces and trouble driving at night. Her past medical and ocular history were unremarkable. Although she denied a family history of ocular or inherited diseases, her sister was having difficulty with night vision and her brother required large print to read. Her medications included daily Vitamin D and Ocuvite supplements. She was born in Sweden, otherwise her social history was insignificant. Her review of systems was negative.
On examination, her best corrected (low myope) Snellen visual acuity measured 20/20 right eye and 20/25 left eye. No afferent pupillary defect was noted, confrontational visual fields were full, and intraocular pressures were 12 mmHg OD and 12 mmHg OS. Anterior segment examination was unremarkable in both eyes. Examination of the right eye was significant areas of RPE atrophy involving the temporal macula, and a few pigment clumps temporal to the macula (Figure 1A). Examination of the left eye was noteworthy for more extensive RPE atrophy involving the fovea and temporal macula (Figure 1B). Optos® wide field fundus autofluorescence (FAF) revealed dense, hypo-autofluorescence in the macula, with surrounding patchy regions of mixed hypo- and reticular-like hyper-autofluorescence involving the entire posterior pole (Figure 2A and B). Spectral domain optical coherence tomography (SD-OCT) showed loss of outer retinal architecture in the macula of both eyes, with extensive photoreceptor layer and RPE atrophy (Figures 3A and B). Fluorescein angiography (FA) of both eyes showed discrete areas of hypofluorescence temporal to the macula with overall increased fluorescence in the macula, but a dark choroid beyond (Figure 4A and B).
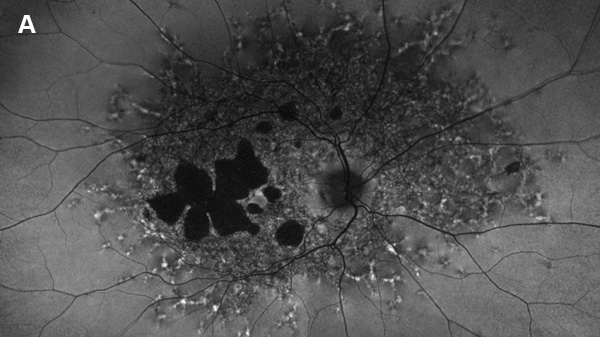
Figure 2A: Fundus autofluorescence of the right eye. Note areas of absent autofluorescence temporal to the fovea and widespread reduced autofluorescence throughout the posterior pole. Small punctate areas of increased autofluorescence are noted beyond.
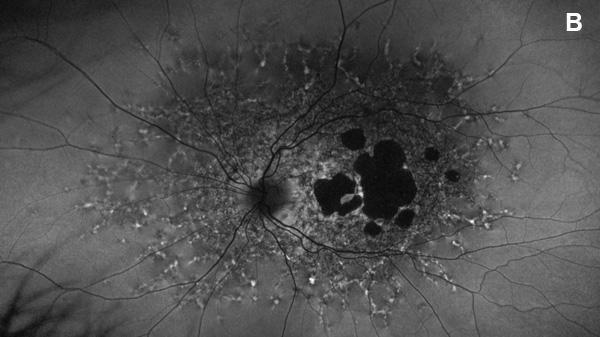
Figure 2B: Fundus autofluorescence of the left eye. Note areas of absent autofluorescence temporal to the fovea and widespread reduced autofluorescence throughout the posterior pole. Small punctate areas of increased autofluorescence are noted beyond.
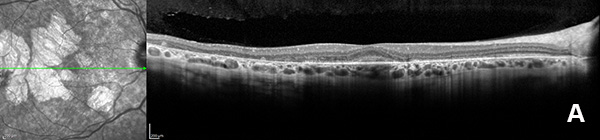
Figure 3A: OCT scan of the right eye. Note the areas of outer retinal atrophy outside the central fovea.
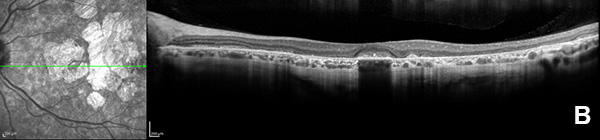
Figure 3B: OCT scan of the left eye. Note the areas of outer retinal atrophy outside the central fovea.
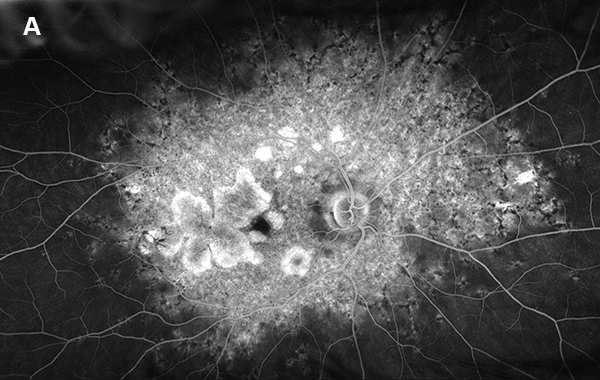
Figure 4A: Wide-field fluorescein angiogram of the right eye. Extensive areas of increased fluorescence is present throughout the posterior pole and discrete atrophy is seen temporal to the macula.

Figure 4B: Wide-field fluorescein angiogram of the left eye. Extensive areas of increased fluorescence is present throughout the posterior pole and a larger area of atrophy is seen in the macula.
Differential Diagnosis
Stargardt disease, pattern Dystrophy, macular degeneration, other macular dystrophy.
Additional History
The patient was observed on an annual basis, and 3 years later, her visual acuity had worsened to 20/30 OD and 20/40 OS. She has noted increased difficulty in reading and seeing large objects in their entirety. Color retinal photos (not shown) and FAF photos confirmed progression of atrophy (Figures 5A and B) optos FAF from 2017 compared to 2/2020). OCT showed sparing of foveal photoreceptors in both eyes, contributing to the relatively preserved visual acuity (Figure 6A and B).
A blood sample was obtained and sent for genetic analysis. The results showed the patient to be heterozygous for two variants in the ABCA4 gene: ABCA4 c.4685T>C, p.(Ile1562THR) and ABCA4 c.768G>T, p.(Val256=), both of which are pathogenic and inherited in a autosomal recessive pattern. Her specific sequence alterations have been known to cause the following phenotypes: cone and rod dystrophy, fundus flavimaculatus, retinal dystrophy, early-onset severe retinitis pigmentosa, and Stargardt disease. Based on her clinical history, examination, and genetic testing, she was formerly diagnosed with Stargardt disease.
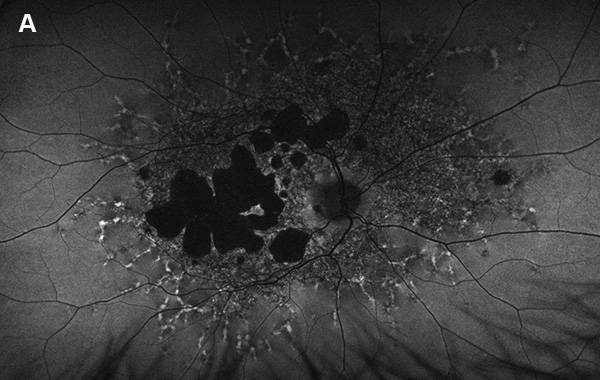
Figure 5A: Wide-field autofluorescence of the right eye. This study is 3 years later from the one shown in Figure 2A. Note the progression of atrophy.
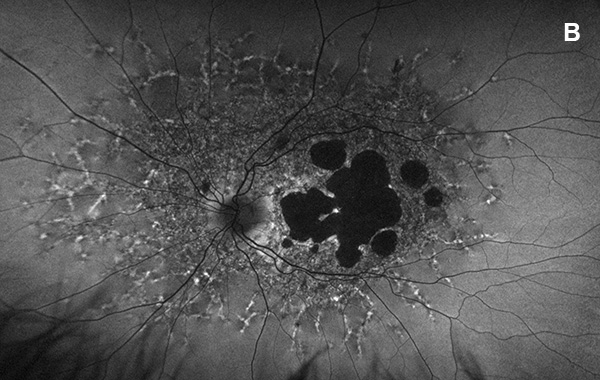
Figure 5A: Wide-field autofluorescence of the left eye. This study is 3 years later from the one shown in Figure 2B. Atrophy has increased in this eye as well.
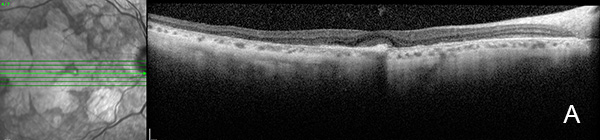
Figure 6A: OCT scan of the right eye. This study is 3 years following the study shown in Figure 3A. Note the areas of outer retinal atrophy with some preservation centrally, consistent with retention of vision.
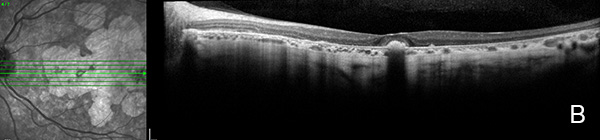
Figure 6A: OCT scan of the left eye. This study is 3 years following the study shown in Figure 3B. Similar to the right eye, there remains some preservation of the central fovea.
Discussion
The ABCA4 gene is the most common location of mutations leading to the phenotypic presentation of Stargardt disease (STGD), which usually results from inheritance in an autosomal recessive pattern. The ABCA4 gene encodes for an ATP-binding cassette membrane protein located in photoreceptor outer segments that facilitates transport of all-trans-retinal aldehyde. The absence or malfunction of this protein leads to the accumulation of bisretinoids, such as A2E, which lead to damage and atrophy of the outer retina and RPE.1 Due to genotypic variability and differences in amount of functioning ABCA4 protein from patient to patient, the clinical signs of STGD can vary widely (Table 1).
Patients with STGD can present at a variety of ages, with earlier presentation (before 20s) typically having a more rapidly-progressive disease course, reaching a visual acuity of 20/200 or worse at a median time of 7 years.2 Patients presenting between ages 21-40 and 41-60 take approximately 22 and 29 years, respectively, to decline to a visual acuity of 20/200 or worse.2 Our 45 year-old patient demonstrated compound heterozygosity for variants in the ABCA4 sequence, which likely explains the late onset of her symptoms and clinical presentation. Despite having later onset STGD, she has already lost 2 lines of vision in both eyes over the course of 3 years and will likely continue this trend based on her ancillary testing which shows minimally intact foveal architecture. The rapidly-progressing nature of her late-onset disease is atypical from what is commonly reported in this age group of patients. Unfortunately, a correlation between her genotype and the severity of her phenotype has yet to be established. While the investigation of genetic therapy for STGD continues, we recommended close observation, the use of UV blocking glasses while outside, and genetic testing within her family.