July, 2023
Presented by Lawrence Chan, MD


Presented by Lawrence Chan, MD
A 69-year-old woman of Japanese descent is referred for retinal pigmentary changes and visual field abnormalities in both eyes.
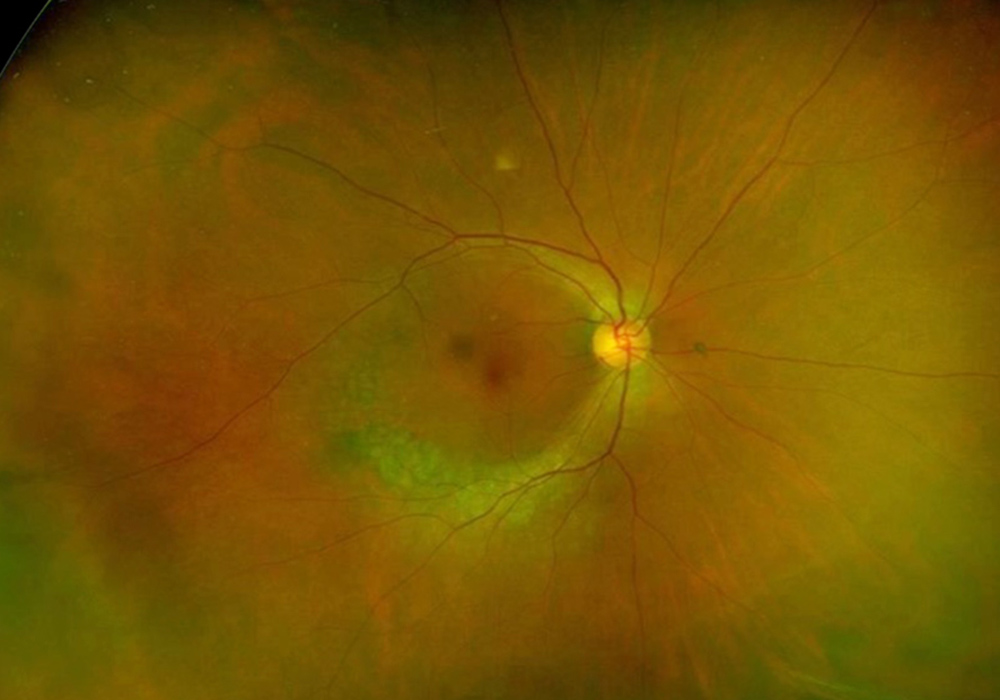
Figure 1A: Wide-field color photo of the right eye. Note the arcuate area of depigmentation along the inferior arcade extending superiorly around the temporal macula.
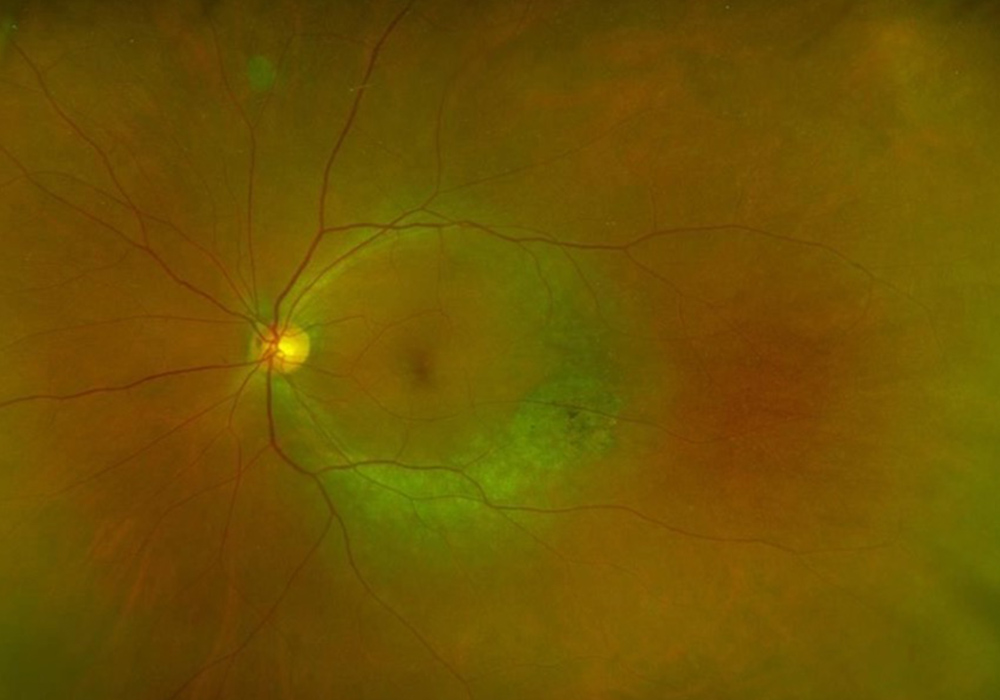
Figure 1B: Wide-field color photo of the left eye. Note the similar pattern of depigmentation as is seen in the right eye.
On presentation, our patient reported no problems with her vision. She denied experiencing any blurriness, distortions, new flashes, floaters, pain, or night blindness. Her past ocular history was notable for myopia and glaucoma suspect status. Her medical history was significant for hypertension, hyperlipidemia, occasional anxiety attacks, and palmoplantar pustulosis. Surgical history was notable for two Cesarean sections approximately 40 years ago. Her medications included atorvastatin, amlodipine, and famotidine as needed, and her supplements included vitamin A, calcium, vitamin D, biotin, glucosamine, and methylsulfonylmethane. Family history was significant for glaucoma in her mother and blindness as a young man in her now deceased brother. She did not have any known drug allergies. She was a former smoker (half a pack a day until she quit 15 years ago). She denied any alcohol or illicit substance use. Review of systems was negative.
The patient's best corrected Snellen visual acuity was 20/40 OD and 20/32 OS. Intraocular pressures were 18 OD and 17 OS. Anterior segment exam was appropriate for age. The posterior segment exam was notable for an arcuate area of RPE attenuation and atrophy along the inferotemporal arcade, extending superiorly to the superotemporal arcade (Figure 1A and B). Fundus autofluorescence imaging showed inferior arcuate areas of hypoautofluorescence corresponding to the partial annular regions of RPE atrophy, as well as subtle inferior perifoveal hyperautofluorescent arcs (Figure 2A and B). OCT macula of both eyes showed perifoveal loss of the outer retinal layers (Figure 3A and B). 24-2 Humphrey visual field testing revealed visual field constriction sparing fixation and predominantly affecting the superior quadrants (Figure 4A and B).
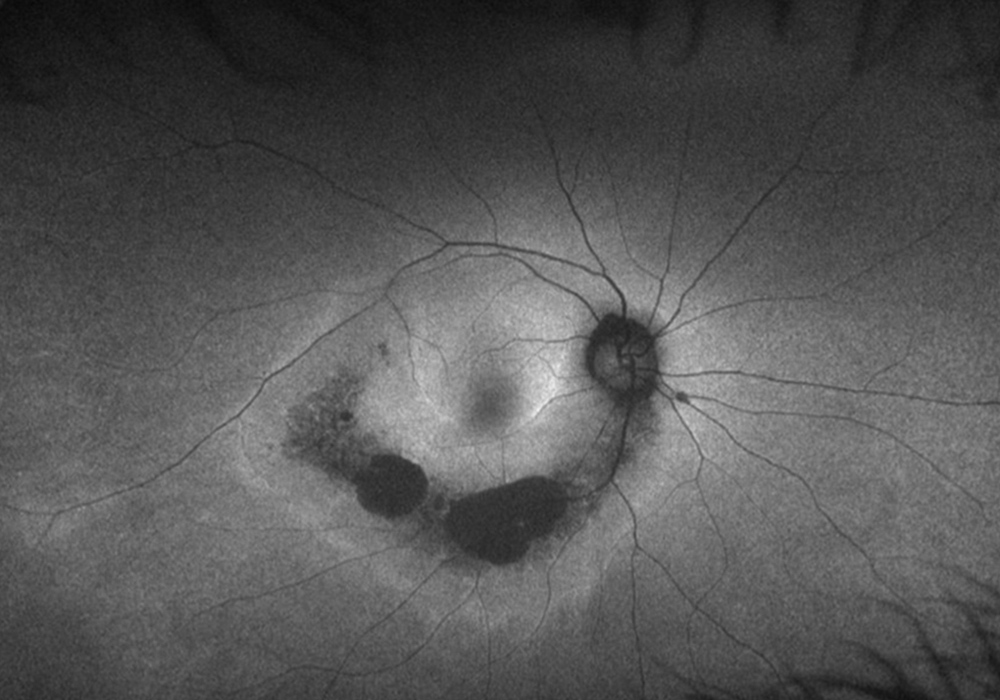
Figure 2A: Wide-field autofluorescence image of the right eye. Note the arcuate area of hypoautofluorescence along the inferotemporal arcade and extending to the temporal macula.
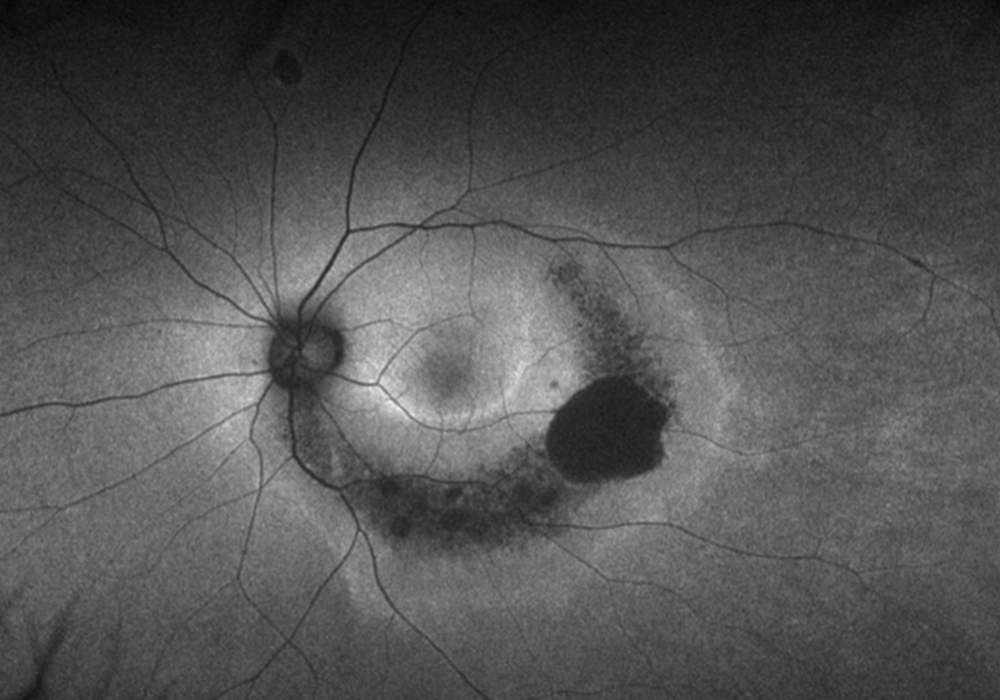
Figure 2B: Wide-field autofluorescence image of the left eye. Note the similar pattern of hypoautofluorescence as is seen in the right eye.
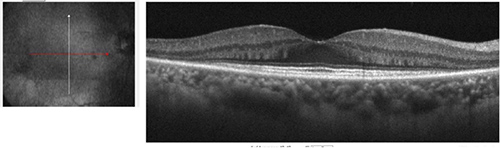
Figure 3A: Horizontal OCT scan of the right macula. Note the perifoveal loss of outer retina.
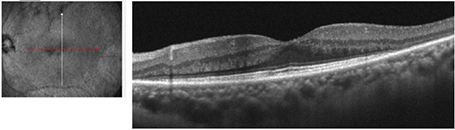
Figure 3B:Horizontal OCT scan of the left macula. A similar pattern of perifoveal loss of outer retina is seen as noted in the right eye.
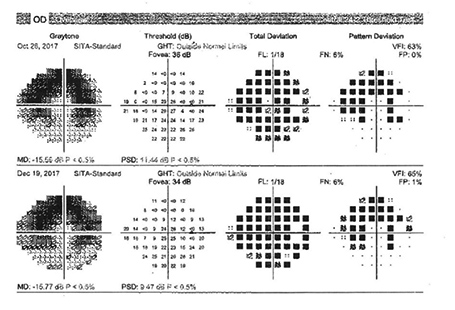
Figure 4A:Automated 24-2 visual field of the right eye showing an arcuate loss that corresponds to the areas of depigmentation noted on the color imaging and the hypoautofluorescence seen on the autofluorescent imaging.
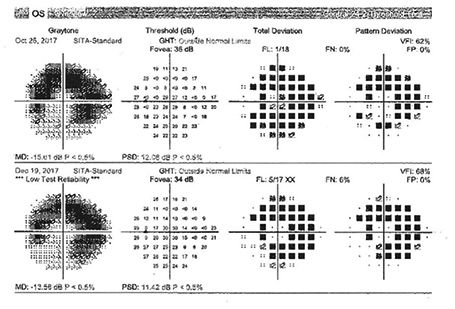
Figure 4B:Automated 24-2 visual field of the left eye showing an arcuate loss that corresponds to the areas of depigmentation noted on the color imaging and the hypoautofluorescence seen on the autofluorescent imaging.
Differential Diagnosis
Additional History and Diagnosis
The patient was followed up annually with mild progression of the areas of inferior arcuate RPE atrophy with preservation of the fovea over a period of five years. Genetic testing with the inherited retinal disorders panel was performed and the patient was found to have two pathogenic variants (c.2528G > A [p.Gly843Glu] and c.8805C > A [p.Tyr2935*]) identified on the Eyes shut homolog (EYS) gene, which is associated with autosomal recessive retinitis pigmentosa
Discussion
Retinitis pigmentosa (RP) refers to a genetically and phenotypically heterogenous group of inherited retinal disorders that affects approximately 1 in 4,000 individuals.1 It is typically characterized by rod, followed by cone photoreceptor degeneration and clinically presents with night blindness and progressive constriction of the visual field.2 RP can occur sporadically or inherited in an autosomal recessive, autosomal dominant, or X-linked manner.
Although RP classically causes bone-spicule hyperpigmentation in the mid-peripheral retina that then moves pericentrally, variants of RP phenotypes have been described, including sector RP (affecting one or two quadrants), pericentral RP (affecting the near-periphery around the vascular arcades), pigmented paravenous RP (pigment accumulation along retinal veins), and RP sine pigmento (absence of bone-spicule pigmentary changes).3-6 Pericentral RP is an uncommon RP subtype with mild disease severity. It has been found to be associated with mutations in RHO, USH2A, HGSNAT, PDE6B, and NR2E3.7,8
EYS, known to be the largest gene expressed in the human eye, has been identified using next-generation sequencing (NGS) as a major causative gene of RP in the Japanese population, accounting for 51% of genetically solved patients in one cohort.9 Our patient, who is of Japanese ancestry, was found to have two pathogenic variants that were previously identified in Japanese patients with non-syndromic autosomal recessive RP. 10 EYS has been found to play an important role in the localization of outer segment proteins and maintenance of actin filaments within the photoreceptors.11 Lo et al. theorize that the affected proteins in patients with EYS mutations cause rod photoreceptors, which reach their highest density along an elliptical ring at the eccentricity of the optic disc (i.e., the vascular arcades), to be more susceptible to stress,4,12 thereby producing a pericentral pattern of RP. Of note, hydroxychloroquine toxicity tends to produce a similar pericentral pattern of changes in patients of Asian ancestry compared to the typical parafoveal/bull's eye pattern seen in patients of other ethnicities, though the exact mechanism for this difference is not clear.13,14